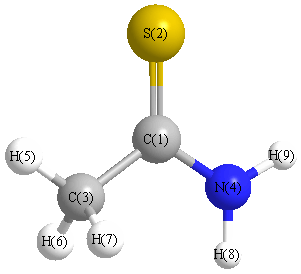
Vibrational Frequencies calculated at MP2/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3756 |
3577 |
51.11 |
|
|
|
| 2 |
A |
3601 |
3430 |
78.11 |
|
|
|
| 3 |
A |
3237 |
3083 |
1.20 |
|
|
|
| 4 |
A |
3164 |
3014 |
9.75 |
|
|
|
| 5 |
A |
3078 |
2932 |
16.65 |
|
|
|
| 6 |
A |
1633 |
1555 |
199.69 |
|
|
|
| 7 |
A |
1485 |
1415 |
31.17 |
|
|
|
| 8 |
A |
1483 |
1413 |
9.51 |
|
|
|
| 9 |
A |
1425 |
1357 |
190.05 |
|
|
|
| 10 |
A |
1396 |
1329 |
53.43 |
|
|
|
| 11 |
A |
1339 |
1275 |
11.34 |
|
|
|
| 12 |
A |
1031 |
982 |
19.86 |
|
|
|
| 13 |
A |
1029 |
980 |
2.72 |
|
|
|
| 14 |
A |
1001 |
954 |
1.48 |
|
|
|
| 15 |
A |
756 |
720 |
2.96 |
|
|
|
| 16 |
A |
618 |
588 |
5.66 |
|
|
|
| 17 |
A |
508 |
484 |
9.08 |
|
|
|
| 18 |
A |
427 |
407 |
0.71 |
|
|
|
| 19 |
A |
380 |
362 |
2.12 |
|
|
|
| 20 |
A |
101 |
96 |
164.39 |
|
|
|
| 21 |
A |
36 |
34 |
8.65 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 15741.2 cm
-1
Scaled (by 0.9525) Zero Point Vibrational Energy (zpe) 14993.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.