Jump to
S1C2
Energy calculated at MP2/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -134.731777 |
| Energy at 298.15K | -134.740037 |
| HF Energy | -134.261331 |
| Nuclear repulsion energy | 82.493791 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3494 |
3328 |
1.24 |
|
|
|
| 2 |
A' |
3170 |
3019 |
33.86 |
|
|
|
| 3 |
A' |
3088 |
2941 |
36.61 |
|
|
|
| 4 |
A' |
3069 |
2923 |
21.72 |
|
|
|
| 5 |
A' |
1658 |
1580 |
16.05 |
|
|
|
| 6 |
A' |
1505 |
1433 |
1.52 |
|
|
|
| 7 |
A' |
1486 |
1415 |
0.08 |
|
|
|
| 8 |
A' |
1409 |
1343 |
12.65 |
|
|
|
| 9 |
A' |
1379 |
1313 |
13.60 |
|
|
|
| 10 |
A' |
1180 |
1124 |
8.23 |
|
|
|
| 11 |
A' |
1106 |
1053 |
30.86 |
|
|
|
| 12 |
A' |
936 |
892 |
120.00 |
|
|
|
| 13 |
A' |
901 |
859 |
21.28 |
|
|
|
| 14 |
A' |
404 |
384 |
5.97 |
|
|
|
| 15 |
A" |
3587 |
3417 |
0.03 |
|
|
|
| 16 |
A" |
3171 |
3020 |
48.37 |
|
|
|
| 17 |
A" |
3138 |
2989 |
9.05 |
|
|
|
| 18 |
A" |
1493 |
1422 |
7.80 |
|
|
|
| 19 |
A" |
1404 |
1337 |
0.03 |
|
|
|
| 20 |
A" |
1279 |
1218 |
0.20 |
|
|
|
| 21 |
A" |
1016 |
968 |
1.58 |
|
|
|
| 22 |
A" |
784 |
746 |
0.66 |
|
|
|
| 23 |
A" |
331 |
315 |
38.40 |
|
|
|
| 24 |
A" |
273 |
260 |
8.81 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20629.1 cm
-1
Scaled (by 0.9525) Zero Point Vibrational Energy (zpe) 19649.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-1.312 |
-0.076 |
0.000 |
| C2 |
0.000 |
0.579 |
0.000 |
| C3 |
1.208 |
-0.363 |
0.000 |
| H4 |
2.160 |
0.195 |
0.000 |
| H5 |
1.195 |
-1.014 |
0.892 |
| H6 |
1.195 |
-1.014 |
-0.892 |
| H7 |
0.046 |
1.241 |
-0.883 |
| H8 |
0.046 |
1.241 |
0.883 |
| H9 |
-1.351 |
-0.706 |
0.808 |
| H10 |
-1.351 |
-0.706 |
-0.808 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
C3 |
H4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.4670 | 2.5363 | 3.4833 | 2.8218 | 2.8218 | 2.0883 | 2.0883 | 1.0247 | 1.0247 |
C2 | 1.4670 | | 1.5316 | 2.1943 | 2.1821 | 2.1821 | 1.1045 | 1.1045 | 2.0317 | 2.0317 | C3 | 2.5363 | 1.5316 | | 1.1039 | 1.1044 | 1.1044 | 2.1682 | 2.1682 | 2.7047 | 2.7047 | H4 | 3.4833 | 2.1943 | 1.1039 | | 1.7854 | 1.7854 | 2.5187 | 2.5187 | 3.7136 | 3.7136 | H5 | 2.8218 | 2.1821 | 1.1044 | 1.7854 | | 1.7844 | 3.0911 | 2.5306 | 2.5659 | 3.0767 | H6 | 2.8218 | 2.1821 | 1.1044 | 1.7854 | 1.7844 | | 2.5306 | 3.0911 | 3.0767 | 2.5659 | H7 | 2.0883 | 1.1045 | 2.1682 | 2.5187 | 3.0911 | 2.5306 | | 1.7660 | 2.9326 | 2.3973 | H8 | 2.0883 | 1.1045 | 2.1682 | 2.5187 | 2.5306 | 3.0911 | 1.7660 | | 2.3973 | 2.9326 | H9 | 1.0247 | 2.0317 | 2.7047 | 3.7136 | 2.5659 | 3.0767 | 2.9326 | 2.3973 | | 1.6155 | H10 | 1.0247 | 2.0317 | 2.7047 | 3.7136 | 3.0767 | 2.5659 | 2.3973 | 2.9326 | 1.6155 | |
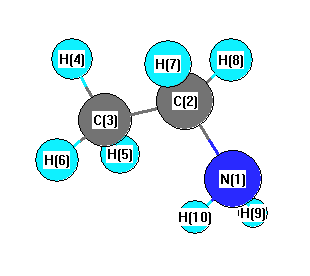 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
C3 |
115.504 |
|
N1 |
C2 |
H7 |
107.773 |
| N1 |
C2 |
H8 |
107.773 |
|
C2 |
N1 |
H9 |
107.935 |
| C2 |
N1 |
H10 |
107.935 |
|
C2 |
C3 |
H4 |
111.705 |
| C2 |
C3 |
H5 |
110.702 |
|
C2 |
C3 |
H6 |
110.702 |
| C3 |
C2 |
H7 |
109.607 |
|
C3 |
C2 |
H8 |
109.607 |
| H4 |
C3 |
H5 |
107.904 |
|
H4 |
C3 |
H6 |
107.904 |
| H5 |
C3 |
H6 |
107.774 |
|
H7 |
C2 |
H8 |
106.155 |
| H9 |
N1 |
H10 |
104.050 |
|
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -134.731218 |
| Energy at 298.15K | |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/cc-pVDZ
Geometric Data calculated at MP2/cc-pVDZ
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability