Jump to
S1C2
S1C3
Energy calculated at MP2/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -1196.110727 |
| Energy at 298.15K | |
| HF Energy | -1194.904022 |
| Nuclear repulsion energy | 379.108897 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3160 |
3000 |
0.19 |
|
|
|
| 2 |
A |
1413 |
1342 |
8.19 |
|
|
|
| 3 |
A |
1309 |
1242 |
9.12 |
|
|
|
| 4 |
A |
1161 |
1102 |
130.11 |
|
|
|
| 5 |
A |
1093 |
1038 |
132.39 |
|
|
|
| 6 |
A |
858 |
814 |
78.03 |
|
|
|
| 7 |
A |
469 |
446 |
0.61 |
|
|
|
| 8 |
A |
315 |
299 |
1.17 |
|
|
|
| 9 |
A |
169 |
161 |
0.51 |
|
|
|
| 10 |
A |
83 |
78 |
0.54 |
|
|
|
| 11 |
B |
3172 |
3012 |
9.12 |
|
|
|
| 12 |
B |
1355 |
1287 |
5.59 |
|
|
|
| 13 |
B |
1247 |
1184 |
24.94 |
|
|
|
| 14 |
B |
1110 |
1054 |
25.18 |
|
|
|
| 15 |
B |
852 |
809 |
90.87 |
|
|
|
| 16 |
B |
440 |
418 |
10.63 |
|
|
|
| 17 |
B |
396 |
376 |
7.08 |
|
|
|
| 18 |
B |
335 |
319 |
10.77 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 9467.3 cm
-1
Scaled (by 0.9495) Zero Point Vibrational Energy (zpe) 8989.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/cc-pVTZ
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.223 |
0.727 |
0.400 |
| C2 |
0.223 |
-0.727 |
0.400 |
| H3 |
-1.305 |
0.806 |
0.353 |
| H4 |
1.305 |
-0.806 |
0.353 |
| F5 |
0.223 |
1.299 |
1.550 |
| F6 |
-0.223 |
-1.299 |
1.550 |
| Cl7 |
0.468 |
1.578 |
-0.982 |
| Cl8 |
-0.468 |
-1.578 |
-0.982 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
H3 |
H4 |
F5 |
F6 |
Cl7 |
Cl8 |
| C1 | | 1.5214 | 1.0865 | 2.1655 | 1.3594 | 2.3301 | 1.7638 | 2.6990 |
C2 | 1.5214 | | 2.1655 | 1.0865 | 2.3301 | 1.3594 | 2.6990 | 1.7638 | H3 | 1.0865 | 2.1655 | | 3.0686 | 2.0030 | 2.6529 | 2.3500 | 2.8579 | H4 | 2.1655 | 1.0865 | 3.0686 | | 2.6529 | 2.0030 | 2.8579 | 2.3500 | F5 | 1.3594 | 2.3301 | 2.0030 | 2.6529 | | 2.6367 | 2.5589 | 3.8945 | F6 | 2.3301 | 1.3594 | 2.6529 | 2.0030 | 2.6367 | | 3.8945 | 2.5589 | Cl7 | 1.7638 | 2.6990 | 2.3500 | 2.8579 | 2.5589 | 3.8945 | | 3.2919 | Cl8 | 2.6990 | 1.7638 | 2.8579 | 2.3500 | 3.8945 | 2.5589 | 3.2919 | |
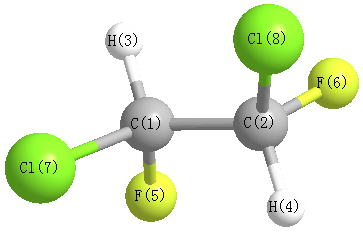 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
H4 |
111.181 |
|
C1 |
C2 |
F6 |
107.835 |
| C1 |
C2 |
Cl8 |
110.266 |
|
C2 |
C1 |
H3 |
111.181 |
| C2 |
C1 |
F5 |
107.835 |
|
C2 |
C1 |
Cl7 |
110.266 |
| H3 |
C1 |
F5 |
109.451 |
|
H3 |
C1 |
Cl7 |
108.736 |
| H4 |
C2 |
F6 |
109.451 |
|
H4 |
C2 |
Cl8 |
108.736 |
| F5 |
C1 |
Cl7 |
109.349 |
|
F6 |
C2 |
Cl8 |
109.349 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C3
Energy calculated at MP2/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -1196.109350 |
| Energy at 298.15K | |
| HF Energy | -1194.903535 |
| Nuclear repulsion energy | 378.848365 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3168 |
3008 |
6.58 |
|
|
|
| 2 |
A |
1388 |
1318 |
3.85 |
|
|
|
| 3 |
A |
1320 |
1254 |
1.89 |
|
|
|
| 4 |
A |
1202 |
1141 |
99.99 |
|
|
|
| 5 |
A |
943 |
895 |
30.39 |
|
|
|
| 6 |
A |
831 |
789 |
0.90 |
|
|
|
| 7 |
A |
419 |
398 |
1.09 |
|
|
|
| 8 |
A |
285 |
271 |
0.49 |
|
|
|
| 9 |
A |
245 |
233 |
0.75 |
|
|
|
| 10 |
A |
82 |
78 |
0.26 |
|
|
|
| 11 |
B |
3162 |
3002 |
4.87 |
|
|
|
| 12 |
B |
1406 |
1335 |
14.73 |
|
|
|
| 13 |
B |
1277 |
1213 |
17.51 |
|
|
|
| 14 |
B |
1118 |
1061 |
118.99 |
|
|
|
| 15 |
B |
808 |
768 |
176.07 |
|
|
|
| 16 |
B |
750 |
712 |
56.13 |
|
|
|
| 17 |
B |
393 |
373 |
4.38 |
|
|
|
| 18 |
B |
185 |
176 |
2.25 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 9491.0 cm
-1
Scaled (by 0.9495) Zero Point Vibrational Energy (zpe) 9011.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/cc-pVTZ
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.218 |
0.728 |
-0.241 |
| C2 |
-0.218 |
-0.728 |
-0.241 |
| H3 |
-0.172 |
1.242 |
-1.115 |
| H4 |
0.172 |
-1.242 |
-1.115 |
| F5 |
-0.218 |
1.347 |
0.881 |
| F6 |
0.218 |
-1.347 |
0.881 |
| Cl7 |
1.986 |
0.800 |
-0.316 |
| Cl8 |
-1.986 |
-0.800 |
-0.316 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
H3 |
H4 |
F5 |
F6 |
Cl7 |
Cl8 |
| C1 | | 1.5195 | 1.0862 | 2.1553 | 1.3527 | 2.3584 | 1.7716 | 2.6827 |
C2 | 1.5195 | | 2.1553 | 1.0862 | 2.3584 | 1.3527 | 2.6827 | 1.7716 | H3 | 1.0862 | 2.1553 | | 2.5067 | 1.9990 | 3.2915 | 2.3432 | 2.8457 | H4 | 2.1553 | 1.0862 | 2.5067 | | 3.2915 | 1.9990 | 2.8457 | 2.3432 | F5 | 1.3527 | 2.3584 | 1.9990 | 3.2915 | | 2.7284 | 2.5665 | 3.0280 | F6 | 2.3584 | 1.3527 | 3.2915 | 1.9990 | 2.7284 | | 3.0280 | 2.5665 | Cl7 | 1.7716 | 2.6827 | 2.3432 | 2.8457 | 2.5665 | 3.0280 | | 4.2824 | Cl8 | 2.6827 | 1.7716 | 2.8457 | 2.3432 | 3.0280 | 2.5665 | 4.2824 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
H4 |
110.509 |
|
C1 |
C2 |
F6 |
110.254 |
| C1 |
C2 |
Cl8 |
108.956 |
|
C2 |
C1 |
H3 |
110.509 |
| C2 |
C1 |
F5 |
110.254 |
|
C2 |
C1 |
Cl7 |
108.956 |
| H3 |
C1 |
F5 |
109.615 |
|
H3 |
C1 |
Cl7 |
107.730 |
| H4 |
C2 |
F6 |
109.615 |
|
H4 |
C2 |
Cl8 |
107.730 |
| F5 |
C1 |
Cl7 |
109.733 |
|
F6 |
C2 |
Cl8 |
109.733 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
Energy calculated at MP2/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -1196.109142 |
| Energy at 298.15K | |
| HF Energy | -1194.902212 |
| Nuclear repulsion energy | 384.935602 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3152 |
2993 |
12.18 |
|
|
|
| 2 |
A |
1395 |
1325 |
13.86 |
|
|
|
| 3 |
A |
1300 |
1234 |
13.24 |
|
|
|
| 4 |
A |
1131 |
1074 |
4.25 |
|
|
|
| 5 |
A |
1086 |
1031 |
48.78 |
|
|
|
| 6 |
A |
710 |
675 |
58.28 |
|
|
|
| 7 |
A |
529 |
502 |
0.18 |
|
|
|
| 8 |
A |
354 |
336 |
1.46 |
|
|
|
| 9 |
A |
180 |
171 |
0.49 |
|
|
|
| 10 |
A |
87 |
83 |
1.00 |
|
|
|
| 11 |
B |
3144 |
2985 |
3.10 |
|
|
|
| 12 |
B |
1351 |
1282 |
41.04 |
|
|
|
| 13 |
B |
1338 |
1270 |
2.77 |
|
|
|
| 14 |
B |
1143 |
1085 |
228.50 |
|
|
|
| 15 |
B |
868 |
824 |
41.42 |
|
|
|
| 16 |
B |
603 |
573 |
47.16 |
|
|
|
| 17 |
B |
401 |
381 |
4.04 |
|
|
|
| 18 |
B |
204 |
193 |
2.17 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 9487.4 cm
-1
Scaled (by 0.9495) Zero Point Vibrational Energy (zpe) 9008.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/cc-pVTZ
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.140 |
0.748 |
0.684 |
| C2 |
0.140 |
-0.748 |
0.684 |
| H3 |
-0.737 |
0.994 |
1.559 |
| H4 |
0.737 |
-0.994 |
1.559 |
| F5 |
1.046 |
1.408 |
0.755 |
| F6 |
-1.046 |
-1.408 |
0.755 |
| Cl7 |
-1.046 |
1.261 |
-0.733 |
| Cl8 |
1.046 |
-1.261 |
-0.733 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
H3 |
H4 |
F5 |
F6 |
Cl7 |
Cl8 |
| C1 | | 1.5223 | 1.0876 | 2.1377 | 1.3590 | 2.3404 | 1.7582 | 2.7295 |
C2 | 1.5223 | | 2.1377 | 1.0876 | 2.3404 | 1.3590 | 2.7295 | 1.7582 | H3 | 1.0876 | 2.1377 | | 2.4751 | 1.9996 | 2.5522 | 2.3277 | 3.6765 | H4 | 2.1377 | 1.0876 | 2.4751 | | 2.5522 | 1.9996 | 3.6765 | 2.3277 | F5 | 1.3590 | 2.3404 | 1.9996 | 2.5522 | | 3.5087 | 2.5712 | 3.0557 | F6 | 2.3404 | 1.3590 | 2.5522 | 1.9996 | 3.5087 | | 3.0557 | 2.5712 | Cl7 | 1.7582 | 2.7295 | 2.3277 | 3.6765 | 2.5712 | 3.0557 | | 3.2770 | Cl8 | 2.7295 | 1.7582 | 3.6765 | 2.3277 | 3.0557 | 2.5712 | 3.2770 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
H4 |
108.845 |
|
C1 |
C2 |
F6 |
108.504 |
| C1 |
C2 |
Cl8 |
112.416 |
|
C2 |
C1 |
H3 |
108.845 |
| C2 |
C1 |
F5 |
108.504 |
|
C2 |
C1 |
Cl7 |
112.416 |
| H3 |
C1 |
F5 |
109.129 |
|
H3 |
C1 |
Cl7 |
107.399 |
| H4 |
C2 |
F6 |
109.129 |
|
H4 |
C2 |
Cl8 |
107.399 |
| F5 |
C1 |
Cl7 |
110.488 |
|
F6 |
C2 |
Cl8 |
110.488 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability