Jump to
S1C2
Energy calculated at MP2/6-311G*
| | hartrees |
|---|
| Energy at 0K | -274.939932 |
| Energy at 298.15K | |
| HF Energy | -274.199664 |
| Nuclear repulsion energy | 117.556804 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2083 |
1979 |
50.90 |
47.95 |
0.12 |
0.22 |
| 2 |
A1 |
788 |
749 |
17.24 |
12.06 |
0.10 |
0.18 |
| 3 |
A1 |
552 |
525 |
55.83 |
1.20 |
0.42 |
0.59 |
| 4 |
A1 |
110 |
105 |
10.35 |
2.23 |
0.69 |
0.82 |
| 5 |
A2 |
469 |
446 |
0.00 |
3.30 |
0.75 |
0.86 |
| 6 |
B1 |
495 |
471 |
92.72 |
0.49 |
0.75 |
0.86 |
| 7 |
B2 |
2086 |
1983 |
868.59 |
5.25 |
0.75 |
0.86 |
| 8 |
B2 |
1184 |
1125 |
107.94 |
1.18 |
0.75 |
0.86 |
| 9 |
B2 |
460 |
437 |
15.21 |
1.58 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 4114.0 cm
-1
Scaled (by 0.9503) Zero Point Vibrational Energy (zpe) 3909.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
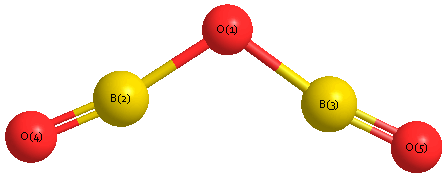
Geometric Data calculated at MP2/6-311G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.662 |
| B2 |
0.000 |
1.210 |
0.089 |
| B3 |
0.000 |
-1.210 |
0.089 |
| O4 |
0.000 |
2.329 |
-0.387 |
| O5 |
0.000 |
-2.329 |
-0.387 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3392 | 1.3392 | 2.5540 | 2.5540 |
B2 | 1.3392 | | 2.4208 | 1.2153 | 3.5710 | B3 | 1.3392 | 2.4208 | | 3.5710 | 1.2153 | O4 | 2.5540 | 1.2153 | 3.5710 | | 4.6575 | O5 | 2.5540 | 3.5710 | 1.2153 | 4.6575 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
177.710 |
|
O1 |
B3 |
O5 |
177.710 |
| B2 |
O1 |
B3 |
129.328 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP2/6-311G*
| | hartrees |
|---|
| Energy at 0K | -274.934833 |
| Energy at 298.15K | |
| HF Energy | -274.198384 |
| Nuclear repulsion energy | 116.343653 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP2/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σg |
2089 |
1985 |
0.00 |
76.56 |
0.16 |
0.28 |
| 2 |
Σg |
667 |
634 |
0.00 |
11.47 |
0.16 |
0.27 |
| 3 |
Σu |
2136 |
2030 |
1401.04 |
0.00 |
0.00 |
0.00 |
| 4 |
Σu |
1304 |
1239 |
118.56 |
0.00 |
0.00 |
0.00 |
| 5 |
Πg |
471 |
447 |
0.00 |
4.17 |
0.75 |
0.86 |
| 5 |
Πg |
471 |
447 |
0.00 |
4.17 |
0.75 |
0.86 |
| 6 |
Πu |
444 |
422 |
112.00 |
0.00 |
0.00 |
0.00 |
| 6 |
Πu |
444 |
422 |
112.00 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
125i |
119i |
1.51 |
0.00 |
0.00 |
0.00 |
| 7 |
Πu |
125i |
119i |
1.51 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 3886.9 cm
-1
Scaled (by 0.9503) Zero Point Vibrational Energy (zpe) 3693.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP2/6-311G*
Point Group is D∞h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.000 |
| B2 |
0.000 |
0.000 |
1.318 |
| B3 |
0.000 |
0.000 |
-1.318 |
| O4 |
0.000 |
0.000 |
2.534 |
| O5 |
0.000 |
0.000 |
-2.534 |
Atom - Atom Distances (Å)
| |
O1 |
B2 |
B3 |
O4 |
O5 |
| O1 | | 1.3177 | 1.3177 | 2.5343 | 2.5343 |
B2 | 1.3177 | | 2.6354 | 1.2166 | 3.8520 | B3 | 1.3177 | 2.6354 | | 3.8520 | 1.2166 | O4 | 2.5343 | 1.2166 | 3.8520 | | 5.0686 | O5 | 2.5343 | 3.8520 | 1.2166 | 5.0686 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
B2 |
O4 |
180.000 |
|
O1 |
B3 |
O5 |
180.000 |
| B2 |
O1 |
B3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability