All results from a given calculation for N(SiH3)3 (trisilylamine)
using model chemistry: MP4/aug-cc-pVTZ
19 10 17 12 22
States and conformations
| State |
Conformation |
minimum conformation |
conformer description |
state description |
| 1 |
1 |
yes |
C3H |
1A' |
Energy calculated at MP4/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -927.310860 |
| Energy at 298.15K | |
| HF Energy | -926.616789 |
| Nuclear repulsion energy | 290.577915 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP4/aug-cc-pVTZ
Geometric Data calculated at MP4/aug-cc-pVTZ
Point Group is C3h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
0.000 |
| Si2 |
0.000 |
1.751 |
0.000 |
| Si3 |
-1.516 |
-0.875 |
0.000 |
| Si4 |
1.516 |
-0.875 |
0.000 |
| H5 |
-1.419 |
2.189 |
0.000 |
| H6 |
-1.186 |
-2.324 |
0.000 |
| H7 |
2.605 |
0.135 |
0.000 |
| H8 |
0.683 |
2.291 |
1.202 |
| H9 |
0.683 |
2.291 |
-1.202 |
| H10 |
-2.326 |
-0.554 |
1.202 |
| H11 |
-2.326 |
-0.554 |
-1.202 |
| H12 |
1.643 |
-1.737 |
1.202 |
| H13 |
1.643 |
-1.737 |
-1.202 |
Atom - Atom Distances (Å)
| |
N1 |
Si2 |
Si3 |
Si4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| N1 | | 1.7507 | 1.7507 | 1.7507 | 2.6086 | 2.6086 | 2.6086 | 2.6761 | 2.6761 | 2.6761 | 2.6761 | 2.6761 | 2.6761 |
Si2 | 1.7507 | | 3.0323 | 3.0323 | 1.4855 | 4.2433 | 3.0654 | 1.4844 | 1.4844 | 3.4880 | 3.4880 | 4.0386 | 4.0386 | Si3 | 1.7507 | 3.0323 | | 3.0323 | 3.0654 | 1.4855 | 4.2433 | 4.0386 | 4.0386 | 1.4844 | 1.4844 | 3.4880 | 3.4880 | Si4 | 1.7507 | 3.0323 | 3.0323 | | 4.2433 | 3.0654 | 1.4855 | 3.4880 | 3.4880 | 4.0386 | 4.0386 | 1.4844 | 1.4844 | H5 | 2.6086 | 1.4855 | 3.0654 | 4.2433 | | 4.5182 | 4.5182 | 2.4241 | 2.4241 | 3.1285 | 3.1285 | 5.1221 | 5.1221 | H6 | 2.6086 | 4.2433 | 1.4855 | 3.0654 | 4.5182 | | 4.5182 | 5.1221 | 5.1221 | 2.4241 | 2.4241 | 3.1285 | 3.1285 | H7 | 2.6086 | 3.0654 | 4.2433 | 1.4855 | 4.5182 | 4.5182 | | 3.1285 | 3.1285 | 5.1221 | 5.1221 | 2.4241 | 2.4241 | H8 | 2.6761 | 1.4844 | 4.0386 | 3.4880 | 2.4241 | 5.1221 | 3.1285 | | 2.4035 | 4.1416 | 4.7884 | 4.1416 | 4.7884 | H9 | 2.6761 | 1.4844 | 4.0386 | 3.4880 | 2.4241 | 5.1221 | 3.1285 | 2.4035 | | 4.7884 | 4.1416 | 4.7884 | 4.1416 | H10 | 2.6761 | 3.4880 | 1.4844 | 4.0386 | 3.1285 | 2.4241 | 5.1221 | 4.1416 | 4.7884 | | 2.4035 | 4.1416 | 4.7884 | H11 | 2.6761 | 3.4880 | 1.4844 | 4.0386 | 3.1285 | 2.4241 | 5.1221 | 4.7884 | 4.1416 | 2.4035 | | 4.7884 | 4.1416 | H12 | 2.6761 | 4.0386 | 3.4880 | 1.4844 | 5.1221 | 3.1285 | 2.4241 | 4.1416 | 4.7884 | 4.1416 | 4.7884 | | 2.4035 | H13 | 2.6761 | 4.0386 | 3.4880 | 1.4844 | 5.1221 | 3.1285 | 2.4241 | 4.7884 | 4.1416 | 4.7884 | 4.1416 | 2.4035 | |
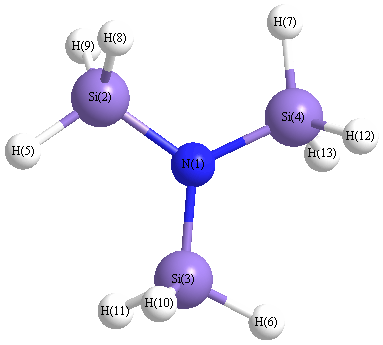 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
Si2 |
H5 |
107.144 |
|
N1 |
Si2 |
H8 |
111.363 |
| N1 |
Si2 |
H9 |
111.363 |
|
N1 |
Si3 |
H6 |
107.144 |
| N1 |
Si3 |
H10 |
111.363 |
|
N1 |
Si3 |
H11 |
111.363 |
| N1 |
Si4 |
H7 |
107.144 |
|
N1 |
Si4 |
H12 |
111.363 |
| N1 |
Si4 |
H13 |
111.363 |
|
Si2 |
N1 |
Si3 |
120.000 |
| Si2 |
N1 |
Si4 |
120.000 |
|
Si3 |
N1 |
Si4 |
120.000 |
| H5 |
Si2 |
H8 |
109.420 |
|
H5 |
Si2 |
H9 |
109.420 |
| H6 |
Si3 |
H10 |
109.420 |
|
H6 |
Si3 |
H11 |
109.420 |
| H7 |
Si4 |
H12 |
109.420 |
|
H7 |
Si4 |
H13 |
109.420 |
| H8 |
Si2 |
H9 |
108.108 |
|
H10 |
Si3 |
H11 |
108.108 |
| H12 |
Si4 |
H13 |
108.108 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability