Jump to
S1C2
S2C1
S2C2
Energy calculated at QCISD/6-31G
| | hartrees |
|---|
| Energy at 0K | -130.882378 |
| Energy at 298.15K | -130.881693 |
| HF Energy | -130.630562 |
| Nuclear repulsion energy | 46.289549 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3315 |
3195 |
8.40 |
|
|
|
| 2 |
A' |
1776 |
1711 |
24.35 |
|
|
|
| 3 |
A' |
1128 |
1087 |
6.56 |
|
|
|
| 4 |
A' |
676 |
651 |
22.24 |
|
|
|
| 5 |
A' |
360 |
347 |
2.23 |
|
|
|
| 6 |
A" |
359 |
346 |
0.14 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3807.0 cm
-1
Scaled (by 0.9636) Zero Point Vibrational Energy (zpe) 3668.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/6-31G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.193 |
-1.246 |
0.000 |
| C2 |
0.000 |
0.092 |
0.000 |
| N3 |
-0.303 |
1.276 |
0.000 |
| H4 |
0.962 |
-2.007 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3525 | 2.5703 | 1.0820 |
C2 | 1.3525 | | 1.2215 | 2.3095 | N3 | 2.5703 | 1.2215 | | 3.5182 | H4 | 1.0820 | 2.3095 | 3.5182 | |
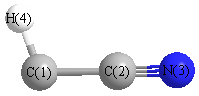 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
173.846 |
|
C2 |
C1 |
H4 |
142.881 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S2C1
S2C2
Energy calculated at QCISD/6-31G
| | hartrees |
|---|
| Energy at 0K | -130.878995 |
| Energy at 298.15K | |
| HF Energy | -130.630003 |
| Nuclear repulsion energy | 46.422961 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
3382 |
3259 |
43.36 |
|
|
|
| 2 |
Σ |
1609 |
1550 |
29.64 |
|
|
|
| 3 |
Σ |
1200 |
1156 |
2.55 |
|
|
|
| 4 |
Π |
333 |
321 |
0.98 |
|
|
|
| 4 |
Π |
333 |
321 |
0.98 |
|
|
|
| 5 |
Π |
584i |
563i |
55.24 |
|
|
|
| 5 |
Π |
584i |
563i |
55.24 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 2844.4 cm
-1
Scaled (by 0.9636) Zero Point Vibrational Energy (zpe) 2740.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/6-31G
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.235 |
| C2 |
0.000 |
0.000 |
0.081 |
| N3 |
0.000 |
0.000 |
1.319 |
| H4 |
0.000 |
0.000 |
-2.312 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3166 | 2.5544 | 1.0763 |
C2 | 1.3166 | | 1.2378 | 2.3930 | N3 | 2.5544 | 1.2378 | | 3.6307 | H4 | 1.0763 | 2.3930 | 3.6307 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
180.000 |
|
C2 |
C1 |
H4 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at QCISD/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.036 |
|
|
|
| 2 |
C |
-0.077 |
|
|
|
| 3 |
N |
-0.190 |
|
|
|
| 4 |
H |
0.303 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-3.351 |
3.351 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
37.406 |
| (<r2>)1/2 |
6.116 |
Jump to
S1C1
S1C2
S2C2
Energy calculated at QCISD/6-31G
| | hartrees |
|---|
| Energy at 0K | -130.840150 |
| Energy at 298.15K | -130.839466 |
| HF Energy | -130.549068 |
| Nuclear repulsion energy | 45.908360 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
2966 |
2858 |
35.80 |
|
|
|
| 2 |
A' |
2005 |
1932 |
29.80 |
|
|
|
| 3 |
A' |
1043 |
1005 |
77.53 |
|
|
|
| 4 |
A' |
906 |
873 |
6.82 |
|
|
|
| 5 |
A' |
374 |
360 |
14.41 |
|
|
|
| 6 |
A" |
287 |
276 |
1.11 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3789.9 cm
-1
Scaled (by 0.9636) Zero Point Vibrational Energy (zpe) 3651.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at QCISD/6-31G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.422 |
-1.246 |
0.000 |
| C2 |
0.000 |
0.110 |
0.000 |
| N3 |
-0.580 |
1.163 |
0.000 |
| H4 |
1.533 |
-1.330 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.4197 | 2.6090 | 1.1150 |
C2 | 1.4197 | | 1.2026 | 2.1036 | N3 | 2.6090 | 1.2026 | | 3.2688 | H4 | 1.1150 | 2.1036 | 3.2688 | |
More geometry information
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S1C2
S2C1
Energy calculated at QCISD/6-31G
| | hartrees |
|---|
| Energy at 0K | -130.840150 |
| Energy at 298.15K | -130.839466 |
| HF Energy | -130.549068 |
| Nuclear repulsion energy | 45.908360 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at QCISD/6-31G
Geometric Data calculated at QCISD/6-31G
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability