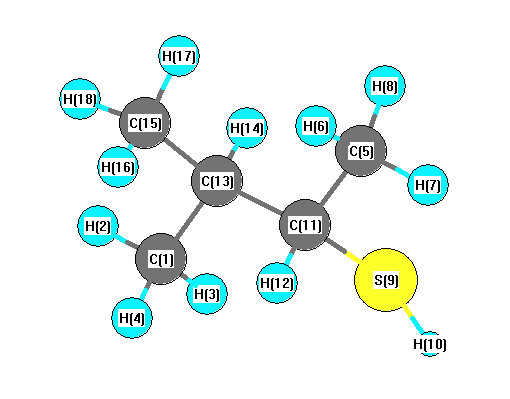
Vibrational Frequencies calculated at LSDA/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3473 |
3110 |
0.30 |
|
|
|
| 2 |
A |
3469 |
3107 |
0.24 |
|
|
|
| 3 |
A |
3466 |
3103 |
0.15 |
|
|
|
| 4 |
A |
3465 |
3103 |
0.27 |
|
|
|
| 5 |
A |
3460 |
3098 |
0.53 |
|
|
|
| 6 |
A |
3457 |
3096 |
0.35 |
|
|
|
| 7 |
A |
3347 |
2997 |
2.58 |
|
|
|
| 8 |
A |
3305 |
2959 |
4.42 |
|
|
|
| 9 |
A |
3297 |
2953 |
1.23 |
|
|
|
| 10 |
A |
3291 |
2947 |
1.93 |
|
|
|
| 11 |
A |
3290 |
2946 |
1.02 |
|
|
|
| 12 |
A |
2944 |
2637 |
22.69 |
|
|
|
| 13 |
A |
1655 |
1482 |
3.46 |
|
|
|
| 14 |
A |
1654 |
1481 |
3.04 |
|
|
|
| 15 |
A |
1645 |
1473 |
2.32 |
|
|
|
| 16 |
A |
1640 |
1469 |
4.24 |
|
|
|
| 17 |
A |
1639 |
1468 |
15.17 |
|
|
|
| 18 |
A |
1629 |
1459 |
4.93 |
|
|
|
| 19 |
A |
1533 |
1373 |
4.14 |
|
|
|
| 20 |
A |
1522 |
1363 |
8.95 |
|
|
|
| 21 |
A |
1513 |
1355 |
7.97 |
|
|
|
| 22 |
A |
1478 |
1323 |
0.99 |
|
|
|
| 23 |
A |
1441 |
1291 |
0.11 |
|
|
|
| 24 |
A |
1408 |
1261 |
4.87 |
|
|
|
| 25 |
A |
1344 |
1203 |
16.32 |
|
|
|
| 26 |
A |
1285 |
1150 |
3.79 |
|
|
|
| 27 |
A |
1265 |
1132 |
11.47 |
|
|
|
| 28 |
A |
1243 |
1113 |
2.80 |
|
|
|
| 29 |
A |
1165 |
1044 |
1.85 |
|
|
|
| 30 |
A |
1127 |
1009 |
14.09 |
|
|
|
| 31 |
A |
1057 |
946 |
10.29 |
|
|
|
| 32 |
A |
1036 |
928 |
0.82 |
|
|
|
| 33 |
A |
1014 |
908 |
5.10 |
|
|
|
| 34 |
A |
1006 |
901 |
4.59 |
|
|
|
| 35 |
A |
1000 |
895 |
3.44 |
|
|
|
| 36 |
A |
884 |
792 |
0.97 |
|
|
|
| 37 |
A |
782 |
700 |
1.19 |
|
|
|
| 38 |
A |
491 |
440 |
0.49 |
|
|
|
| 39 |
A |
406 |
364 |
0.15 |
|
|
|
| 40 |
A |
370 |
331 |
0.18 |
|
|
|
| 41 |
A |
343 |
307 |
0.75 |
|
|
|
| 42 |
A |
340 |
305 |
0.63 |
|
|
|
| 43 |
A |
246 |
221 |
7.68 |
|
|
|
| 44 |
A |
241 |
216 |
4.14 |
|
|
|
| 45 |
A |
216 |
194 |
0.03 |
|
|
|
| 46 |
A |
206 |
185 |
2.15 |
|
|
|
| 47 |
A |
198 |
178 |
2.80 |
|
|
|
| 48 |
A |
70 |
63 |
0.58 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 38177.8 cm
-1
Scaled (by 0.8955) Zero Point Vibrational Energy (zpe) 34188.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.278 |
|
|
|
| 2 |
H |
0.090 |
|
|
|
| 3 |
H |
0.096 |
|
|
|
| 4 |
H |
0.089 |
|
|
|
| 5 |
C |
-0.279 |
|
|
|
| 6 |
H |
0.094 |
|
|
|
| 7 |
H |
0.089 |
|
|
|
| 8 |
H |
0.095 |
|
|
|
| 9 |
S |
0.047 |
|
|
|
| 10 |
H |
0.002 |
|
|
|
| 11 |
C |
-0.148 |
|
|
|
| 12 |
H |
0.087 |
|
|
|
| 13 |
C |
-0.072 |
|
|
|
| 14 |
H |
0.089 |
|
|
|
| 15 |
C |
-0.275 |
|
|
|
| 16 |
H |
0.089 |
|
|
|
| 17 |
H |
0.091 |
|
|
|
| 18 |
H |
0.093 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.230 |
0.899 |
0.517 |
1.062 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.805 |
1.557 |
1.296 |
| y |
1.557 |
-43.522 |
0.304 |
| z |
1.296 |
0.304 |
-42.539 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.225 |
1.557 |
1.296 |
| y |
1.557 |
-1.350 |
0.304 |
| z |
1.296 |
0.304 |
0.124 |
|
| Polar |
|---|
| 3z2-r2 | 0.249 |
|---|
| x2-y2 | 1.717 |
|---|
| xy | 1.557 |
|---|
| xz | 1.296 |
|---|
| yz | 0.304 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.135 |
-0.053 |
0.193 |
| y |
-0.053 |
4.962 |
0.359 |
| z |
0.193 |
0.359 |
3.993 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |