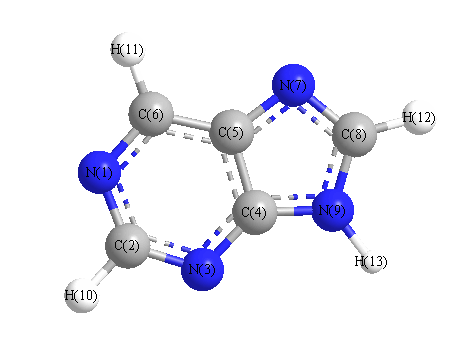
Vibrational Frequencies calculated at LSDA/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3571 |
3504 |
90.50 |
|
|
|
| 2 |
A' |
3181 |
3121 |
0.23 |
|
|
|
| 3 |
A' |
3109 |
3051 |
11.82 |
|
|
|
| 4 |
A' |
3097 |
3039 |
11.32 |
|
|
|
| 5 |
A' |
1662 |
1631 |
56.60 |
|
|
|
| 6 |
A' |
1615 |
1585 |
62.30 |
|
|
|
| 7 |
A' |
1519 |
1491 |
29.01 |
|
|
|
| 8 |
A' |
1482 |
1454 |
3.21 |
|
|
|
| 9 |
A' |
1436 |
1409 |
55.77 |
|
|
|
| 10 |
A' |
1392 |
1366 |
10.63 |
|
|
|
| 11 |
A' |
1376 |
1351 |
76.32 |
|
|
|
| 12 |
A' |
1348 |
1323 |
20.48 |
|
|
|
| 13 |
A' |
1295 |
1271 |
3.14 |
|
|
|
| 14 |
A' |
1265 |
1242 |
20.87 |
|
|
|
| 15 |
A' |
1188 |
1166 |
10.38 |
|
|
|
| 16 |
A' |
1134 |
1112 |
5.65 |
|
|
|
| 17 |
A' |
1066 |
1046 |
11.91 |
|
|
|
| 18 |
A' |
926 |
908 |
2.10 |
|
|
|
| 19 |
A' |
893 |
876 |
10.68 |
|
|
|
| 20 |
A' |
803 |
788 |
14.13 |
|
|
|
| 21 |
A' |
646 |
634 |
0.66 |
|
|
|
| 22 |
A' |
557 |
547 |
4.91 |
|
|
|
| 23 |
A' |
436 |
427 |
13.08 |
|
|
|
| 24 |
A" |
951 |
933 |
0.54 |
|
|
|
| 25 |
A" |
905 |
888 |
10.09 |
|
|
|
| 26 |
A" |
838 |
823 |
3.94 |
|
|
|
| 27 |
A" |
796 |
781 |
10.31 |
|
|
|
| 28 |
A" |
656 |
644 |
4.46 |
|
|
|
| 29 |
A" |
612 |
601 |
29.35 |
|
|
|
| 30 |
A" |
562 |
551 |
96.95 |
|
|
|
| 31 |
A" |
401 |
393 |
0.95 |
|
|
|
| 32 |
A" |
236 |
232 |
0.32 |
|
|
|
| 33 |
A" |
224 |
220 |
4.31 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20588.4 cm
-1
Scaled (by 0.9813) Zero Point Vibrational Energy (zpe) 20203.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.367 |
|
|
|
| 2 |
C |
0.126 |
|
|
|
| 3 |
N |
-0.403 |
|
|
|
| 4 |
C |
0.433 |
|
|
|
| 5 |
C |
0.200 |
|
|
|
| 6 |
C |
0.037 |
|
|
|
| 7 |
N |
-0.447 |
|
|
|
| 8 |
C |
0.213 |
|
|
|
| 9 |
N |
-0.543 |
|
|
|
| 10 |
H |
0.140 |
|
|
|
| 11 |
H |
0.148 |
|
|
|
| 12 |
H |
0.161 |
|
|
|
| 13 |
H |
0.303 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.218 |
3.042 |
0.000 |
3.765 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-49.700 |
6.115 |
0.000 |
| y |
6.115 |
-46.239 |
0.000 |
| z |
0.000 |
0.000 |
-51.097 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.032 |
6.115 |
0.000 |
| y |
6.115 |
4.159 |
0.000 |
| z |
0.000 |
0.000 |
-3.128 |
|
| Polar |
|---|
| 3z2-r2 | -6.255 |
|---|
| x2-y2 | -3.461 |
|---|
| xy | 6.115 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
15.125 |
1.377 |
0.000 |
| y |
1.377 |
11.585 |
0.000 |
| z |
0.000 |
0.000 |
4.275 |
<r2> (average value of r
2) Å
2
| <r2> |
252.213 |
| (<r2>)1/2 |
15.881 |