Jump to
S1C2
Vibrational Frequencies calculated at LSDA/6-31G**
Geometric Data calculated at LSDA/6-31G**
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at LSDA/6-31G**
| | hartrees |
|---|
| Energy at 0K | -622.889575 |
| Energy at 298.15K | -622.893278 |
| HF Energy | -622.889575 |
| Nuclear repulsion energy | 191.735956 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at LSDA/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3592 |
3524 |
83.86 |
|
|
|
| 2 |
A' |
1226 |
1203 |
142.99 |
|
|
|
| 3 |
A' |
1070 |
1050 |
4.95 |
|
|
|
| 4 |
A' |
765 |
750 |
130.96 |
|
|
|
| 5 |
A' |
467 |
459 |
67.56 |
|
|
|
| 6 |
A' |
423 |
415 |
103.08 |
|
|
|
| 7 |
A' |
291 |
286 |
17.30 |
|
|
|
| 8 |
A" |
3588 |
3521 |
27.28 |
|
|
|
| 9 |
A" |
1025 |
1006 |
62.13 |
|
|
|
| 10 |
A" |
773 |
759 |
334.80 |
|
|
|
| 11 |
A" |
432 |
424 |
76.50 |
|
|
|
| 12 |
A" |
171 |
168 |
11.10 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 6911.5 cm
-1
Scaled (by 0.9813) Zero Point Vibrational Energy (zpe) 6782.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at LSDA/6-31G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.302 |
0.342 |
0.000 |
| O2 |
-1.048 |
0.944 |
0.000 |
| O3 |
0.302 |
-0.703 |
1.273 |
| O4 |
0.302 |
-0.703 |
-1.273 |
| H5 |
-0.646 |
-0.889 |
1.474 |
| H6 |
-0.646 |
-0.889 |
-1.474 |
Atom - Atom Distances (Å)
| |
S1 |
O2 |
O3 |
O4 |
H5 |
H6 |
| S1 | | 1.4788 | 1.6476 | 1.6476 | 2.1415 | 2.1415 |
O2 | 1.4788 | | 2.4819 | 2.4819 | 2.3856 | 2.3856 | O3 | 1.6476 | 2.4819 | | 2.5470 | 0.9871 | 2.9121 | O4 | 1.6476 | 2.4819 | 2.5470 | | 2.9121 | 0.9871 | H5 | 2.1415 | 2.3856 | 0.9871 | 2.9121 | | 2.9471 | H6 | 2.1415 | 2.3856 | 2.9121 | 0.9871 | 2.9471 | |
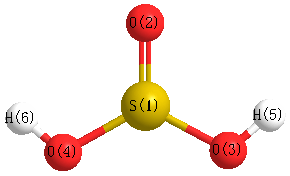 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
O3 |
H5 |
106.012 |
|
S1 |
O4 |
H6 |
106.012 |
| O2 |
S1 |
O3 |
104.962 |
|
O2 |
S1 |
O4 |
104.962 |
| O3 |
S1 |
O4 |
101.235 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.901 |
|
|
|
| 2 |
O |
-0.527 |
|
|
|
| 3 |
O |
-0.526 |
|
|
|
| 4 |
O |
-0.526 |
|
|
|
| 5 |
H |
0.340 |
|
|
|
| 6 |
H |
0.340 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.140 |
-1.331 |
0.000 |
1.752 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.934 |
5.262 |
0.000 |
| y |
5.262 |
-29.995 |
0.000 |
| z |
0.000 |
0.000 |
-28.155 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.858 |
5.262 |
0.000 |
| y |
5.262 |
-0.950 |
0.000 |
| z |
0.000 |
0.000 |
1.809 |
|
| Polar |
|---|
| 3z2-r2 | 3.618 |
|---|
| x2-y2 | 0.061 |
|---|
| xy | 5.262 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.928 |
-0.260 |
0.000 |
| y |
-0.260 |
3.861 |
0.000 |
| z |
0.000 |
0.000 |
4.439 |
<r2> (average value of r
2) Å
2
| <r2> |
79.677 |
| (<r2>)1/2 |
8.926 |