Jump to
S1C2
Energy calculated at LSDA/6-31G**
| | hartrees |
|---|
| Energy at 0K | -340.655317 |
| Energy at 298.15K | |
| HF Energy | -340.655317 |
| Nuclear repulsion energy | 246.229784 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at LSDA/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2985 |
2929 |
48.05 |
|
|
|
| 2 |
A1 |
1957 |
1920 |
524.20 |
|
|
|
| 3 |
A1 |
1493 |
1465 |
1.33 |
|
|
|
| 4 |
A1 |
1343 |
1318 |
0.38 |
|
|
|
| 5 |
A1 |
1221 |
1198 |
212.05 |
|
|
|
| 6 |
A1 |
953 |
935 |
4.79 |
|
|
|
| 7 |
A1 |
853 |
837 |
25.20 |
|
|
|
| 8 |
A1 |
718 |
705 |
0.42 |
|
|
|
| 9 |
A2 |
3015 |
2958 |
0.00 |
|
|
|
| 10 |
A2 |
1200 |
1177 |
0.00 |
|
|
|
| 11 |
A2 |
1126 |
1105 |
0.00 |
|
|
|
| 12 |
A2 |
183i |
179i |
0.00 |
|
|
|
| 13 |
B1 |
3035 |
2978 |
32.01 |
|
|
|
| 14 |
B1 |
1225 |
1202 |
0.24 |
|
|
|
| 15 |
B1 |
838 |
822 |
0.19 |
|
|
|
| 16 |
B1 |
730 |
717 |
16.15 |
|
|
|
| 17 |
B1 |
155 |
152 |
1.47 |
|
|
|
| 18 |
B2 |
2981 |
2925 |
22.17 |
|
|
|
| 19 |
B2 |
1480 |
1452 |
5.66 |
|
|
|
| 20 |
B2 |
1356 |
1331 |
15.14 |
|
|
|
| 21 |
B2 |
1209 |
1186 |
15.48 |
|
|
|
| 22 |
B2 |
1054 |
1034 |
287.64 |
|
|
|
| 23 |
B2 |
745 |
731 |
0.48 |
|
|
|
| 24 |
B2 |
497 |
487 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 15992.0 cm
-1
Scaled (by 0.9813) Zero Point Vibrational Energy (zpe) 15692.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at LSDA/6-31G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.860 |
| O2 |
0.000 |
0.000 |
2.050 |
| O3 |
0.000 |
1.123 |
0.056 |
| O4 |
0.000 |
-1.123 |
0.056 |
| C5 |
0.000 |
0.771 |
-1.279 |
| C6 |
0.000 |
-0.771 |
-1.279 |
| H7 |
-0.894 |
1.189 |
-1.776 |
| H8 |
0.894 |
1.189 |
-1.776 |
| H9 |
0.894 |
-1.189 |
-1.776 |
| H10 |
-0.894 |
-1.189 |
-1.776 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1903 | 1.3812 | 1.3812 | 2.2740 | 2.2740 | 3.0271 | 3.0271 | 3.0271 | 3.0271 |
O2 | 1.1903 | | 2.2887 | 2.2887 | 3.4177 | 3.4177 | 4.1056 | 4.1056 | 4.1056 | 4.1056 | O3 | 1.3812 | 2.2887 | | 2.2460 | 1.3812 | 2.3170 | 2.0399 | 2.0399 | 3.0824 | 3.0824 | O4 | 1.3812 | 2.2887 | 2.2460 | | 2.3170 | 1.3812 | 3.0824 | 3.0824 | 2.0399 | 2.0399 | C5 | 2.2740 | 3.4177 | 1.3812 | 2.3170 | | 1.5410 | 1.1051 | 1.1051 | 2.2102 | 2.2102 | C6 | 2.2740 | 3.4177 | 2.3170 | 1.3812 | 1.5410 | | 2.2102 | 2.2102 | 1.1051 | 1.1051 | H7 | 3.0271 | 4.1056 | 2.0399 | 3.0824 | 1.1051 | 2.2102 | | 1.7881 | 2.9749 | 2.3775 | H8 | 3.0271 | 4.1056 | 2.0399 | 3.0824 | 1.1051 | 2.2102 | 1.7881 | | 2.3775 | 2.9749 | H9 | 3.0271 | 4.1056 | 3.0824 | 2.0399 | 2.2102 | 1.1051 | 2.9749 | 2.3775 | | 1.7881 | H10 | 3.0271 | 4.1056 | 3.0824 | 2.0399 | 2.2102 | 1.1051 | 2.3775 | 2.9749 | 1.7881 | |
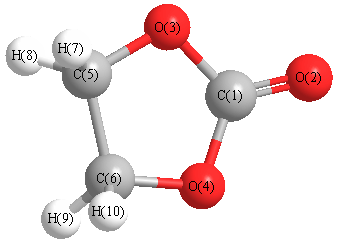 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
110.817 |
|
C1 |
O4 |
C6 |
110.817 |
| O2 |
C1 |
O3 |
125.602 |
|
O2 |
C1 |
O4 |
125.602 |
| O3 |
C1 |
O4 |
108.796 |
|
O3 |
C5 |
C6 |
104.785 |
| O3 |
C5 |
H7 |
109.768 |
|
O3 |
C5 |
H8 |
109.768 |
| O4 |
C6 |
C5 |
104.785 |
|
O4 |
C6 |
H9 |
109.768 |
| O4 |
C6 |
H10 |
109.768 |
|
C5 |
C6 |
H9 |
112.237 |
| C5 |
C6 |
H10 |
112.237 |
|
C6 |
C5 |
H7 |
112.237 |
| C6 |
C5 |
H8 |
112.237 |
|
H7 |
C5 |
H8 |
108.007 |
| H9 |
C6 |
H10 |
108.007 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.658 |
|
|
|
| 2 |
O |
-0.381 |
|
|
|
| 3 |
O |
-0.379 |
|
|
|
| 4 |
O |
-0.379 |
|
|
|
| 5 |
C |
-0.083 |
|
|
|
| 6 |
C |
-0.083 |
|
|
|
| 7 |
H |
0.162 |
|
|
|
| 8 |
H |
0.162 |
|
|
|
| 9 |
H |
0.162 |
|
|
|
| 10 |
H |
0.162 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-4.734 |
4.734 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.914 |
0.000 |
0.000 |
| y |
0.000 |
-35.606 |
0.000 |
| z |
0.000 |
0.000 |
-35.279 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.528 |
0.000 |
0.000 |
| y |
0.000 |
-2.009 |
0.000 |
| z |
0.000 |
0.000 |
-1.519 |
|
| Polar |
|---|
| 3z2-r2 | -3.038 |
|---|
| x2-y2 | 3.692 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.309 |
0.000 |
0.000 |
| y |
0.000 |
5.201 |
0.000 |
| z |
0.000 |
0.000 |
7.538 |
<r2> (average value of r
2) Å
2
| <r2> |
127.937 |
| (<r2>)1/2 |
11.311 |
Jump to
S1C1
Energy calculated at LSDA/6-31G**
| | hartrees |
|---|
| Energy at 0K | -340.656460 |
| Energy at 298.15K | -340.662843 |
| HF Energy | -340.656460 |
| Nuclear repulsion energy | 246.946441 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at LSDA/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3052 |
2995 |
13.10 |
|
|
|
| 2 |
A |
2959 |
2903 |
24.98 |
|
|
|
| 3 |
A |
1959 |
1922 |
505.24 |
|
|
|
| 4 |
A |
1475 |
1447 |
1.83 |
|
|
|
| 5 |
A |
1350 |
1325 |
7.32 |
|
|
|
| 6 |
A |
1220 |
1197 |
125.91 |
|
|
|
| 7 |
A |
1205 |
1183 |
58.05 |
|
|
|
| 8 |
A |
1114 |
1093 |
16.72 |
|
|
|
| 9 |
A |
955 |
937 |
0.92 |
|
|
|
| 10 |
A |
846 |
830 |
28.50 |
|
|
|
| 11 |
A |
716 |
702 |
0.58 |
|
|
|
| 12 |
A |
165 |
162 |
0.59 |
|
|
|
| 13 |
B |
3059 |
3002 |
15.16 |
|
|
|
| 14 |
B |
2967 |
2912 |
43.02 |
|
|
|
| 15 |
B |
1468 |
1440 |
8.04 |
|
|
|
| 16 |
B |
1349 |
1324 |
12.56 |
|
|
|
| 17 |
B |
1235 |
1212 |
3.22 |
|
|
|
| 18 |
B |
1169 |
1147 |
9.44 |
|
|
|
| 19 |
B |
1045 |
1025 |
267.32 |
|
|
|
| 20 |
B |
890 |
873 |
3.27 |
|
|
|
| 21 |
B |
744 |
730 |
17.81 |
|
|
|
| 22 |
B |
664 |
652 |
0.25 |
|
|
|
| 23 |
B |
497 |
488 |
0.08 |
|
|
|
| 24 |
B |
163 |
160 |
1.30 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16130.7 cm
-1
Scaled (by 0.9813) Zero Point Vibrational Energy (zpe) 15829.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at LSDA/6-31G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.854 |
| O2 |
0.000 |
0.000 |
2.044 |
| O3 |
0.000 |
1.124 |
0.043 |
| O4 |
0.000 |
-1.124 |
0.043 |
| C5 |
0.213 |
0.731 |
-1.268 |
| C6 |
-0.213 |
-0.731 |
-1.268 |
| H7 |
-0.384 |
1.356 |
-1.950 |
| H8 |
1.285 |
0.846 |
-1.527 |
| H9 |
0.384 |
-1.356 |
-1.950 |
| H10 |
-1.285 |
-0.846 |
-1.527 |
Atom - Atom Distances (Å)
| |
C1 |
O2 |
O3 |
O4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
| C1 | | 1.1900 | 1.3853 | 1.3853 | 2.2538 | 2.2538 | 3.1386 | 2.8341 | 3.1386 | 2.8341 |
O2 | 1.1900 | | 2.2942 | 2.2942 | 3.3978 | 3.3978 | 4.2357 | 3.8876 | 4.2357 | 3.8876 | O3 | 1.3853 | 2.2942 | | 2.2471 | 1.3853 | 2.2809 | 2.0439 | 2.0476 | 3.2052 | 2.8274 | O4 | 1.3853 | 2.2942 | 2.2471 | | 2.2809 | 1.3853 | 3.2052 | 2.8274 | 2.0439 | 2.0476 | C5 | 2.2538 | 3.3978 | 1.3853 | 2.2809 | | 1.5220 | 1.1020 | 1.1085 | 2.2025 | 2.1898 | C6 | 2.2538 | 3.3978 | 2.2809 | 1.3853 | 1.5220 | | 2.2025 | 2.1898 | 1.1020 | 1.1085 | H7 | 3.1386 | 4.2357 | 2.0439 | 3.2052 | 1.1020 | 2.2025 | | 1.7958 | 2.8195 | 2.4168 | H8 | 2.8341 | 3.8876 | 2.0476 | 2.8274 | 1.1085 | 2.1898 | 1.7958 | | 2.4168 | 3.0762 | H9 | 3.1386 | 4.2357 | 3.2052 | 2.0439 | 2.2025 | 1.1020 | 2.8195 | 2.4168 | | 1.7958 | H10 | 2.8341 | 3.8876 | 2.8274 | 2.0476 | 2.1898 | 1.1085 | 2.4168 | 3.0762 | 1.7958 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O3 |
C5 |
108.876 |
|
C1 |
O4 |
C6 |
108.876 |
| O2 |
C1 |
O3 |
125.798 |
|
O2 |
C1 |
O4 |
125.798 |
| O3 |
C1 |
O4 |
108.405 |
|
O3 |
C5 |
C6 |
103.256 |
| O3 |
C5 |
H7 |
109.998 |
|
O3 |
C5 |
H8 |
109.895 |
| O4 |
C6 |
C5 |
103.256 |
|
O4 |
C6 |
H9 |
109.998 |
| O4 |
C6 |
H10 |
109.895 |
|
C5 |
C6 |
H9 |
113.175 |
| C5 |
C6 |
H10 |
111.746 |
|
C6 |
C5 |
H7 |
113.175 |
| C6 |
C5 |
H8 |
111.746 |
|
H7 |
C5 |
H8 |
108.662 |
| H9 |
C6 |
H10 |
108.662 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.664 |
|
|
|
| 2 |
O |
-0.382 |
|
|
|
| 3 |
O |
-0.380 |
|
|
|
| 4 |
O |
-0.380 |
|
|
|
| 5 |
C |
-0.085 |
|
|
|
| 6 |
C |
-0.085 |
|
|
|
| 7 |
H |
0.164 |
|
|
|
| 8 |
H |
0.159 |
|
|
|
| 9 |
H |
0.164 |
|
|
|
| 10 |
H |
0.159 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-4.665 |
4.665 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.991 |
0.227 |
0.000 |
| y |
0.227 |
-35.633 |
0.000 |
| z |
0.000 |
0.000 |
-35.063 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.357 |
0.227 |
0.000 |
| y |
0.227 |
-2.106 |
0.000 |
| z |
0.000 |
0.000 |
-1.251 |
|
| Polar |
|---|
| 3z2-r2 | -2.503 |
|---|
| x2-y2 | 3.642 |
|---|
| xy | 0.227 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.420 |
0.166 |
0.000 |
| y |
0.166 |
5.131 |
0.000 |
| z |
0.000 |
0.000 |
7.494 |
<r2> (average value of r
2) Å
2
| <r2> |
126.606 |
| (<r2>)1/2 |
11.252 |