Vibrational Frequencies calculated at LSDA/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3056 |
3022 |
15.93 |
|
|
|
| 2 |
A' |
3051 |
3017 |
17.09 |
|
|
|
| 3 |
A' |
2970 |
2938 |
31.66 |
|
|
|
| 4 |
A' |
2963 |
2930 |
8.37 |
|
|
|
| 5 |
A' |
2881 |
2849 |
70.32 |
|
|
|
| 6 |
A' |
2840 |
2809 |
48.31 |
|
|
|
| 7 |
A' |
1452 |
1436 |
8.25 |
|
|
|
| 8 |
A' |
1432 |
1417 |
12.45 |
|
|
|
| 9 |
A' |
1424 |
1409 |
3.87 |
|
|
|
| 10 |
A' |
1414 |
1398 |
4.12 |
|
|
|
| 11 |
A' |
1403 |
1388 |
2.20 |
|
|
|
| 12 |
A' |
1361 |
1346 |
11.03 |
|
|
|
| 13 |
A' |
1344 |
1329 |
7.73 |
|
|
|
| 14 |
A' |
1271 |
1257 |
3.06 |
|
|
|
| 15 |
A' |
1208 |
1195 |
155.83 |
|
|
|
| 16 |
A' |
1177 |
1165 |
22.97 |
|
|
|
| 17 |
A' |
1106 |
1094 |
5.19 |
|
|
|
| 18 |
A' |
1068 |
1056 |
3.89 |
|
|
|
| 19 |
A' |
1012 |
1001 |
23.51 |
|
|
|
| 20 |
A' |
891 |
881 |
9.16 |
|
|
|
| 21 |
A' |
440 |
436 |
0.83 |
|
|
|
| 22 |
A' |
402 |
397 |
3.02 |
|
|
|
| 23 |
A' |
186 |
184 |
1.16 |
|
|
|
| 24 |
A" |
3037 |
3004 |
30.57 |
|
|
|
| 25 |
A" |
3007 |
2975 |
4.04 |
|
|
|
| 26 |
A" |
2924 |
2893 |
46.80 |
|
|
|
| 27 |
A" |
2862 |
2831 |
67.63 |
|
|
|
| 28 |
A" |
1423 |
1407 |
11.06 |
|
|
|
| 29 |
A" |
1402 |
1387 |
10.88 |
|
|
|
| 30 |
A" |
1251 |
1237 |
0.29 |
|
|
|
| 31 |
A" |
1210 |
1197 |
2.61 |
|
|
|
| 32 |
A" |
1152 |
1139 |
4.48 |
|
|
|
| 33 |
A" |
1125 |
1113 |
0.06 |
|
|
|
| 34 |
A" |
863 |
854 |
1.91 |
|
|
|
| 35 |
A" |
739 |
731 |
3.19 |
|
|
|
| 36 |
A" |
242 |
240 |
2.31 |
|
|
|
| 37 |
A" |
238 |
235 |
0.30 |
|
|
|
| 38 |
A" |
114 |
113 |
2.66 |
|
|
|
| 39 |
A" |
106 |
105 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 29022.3 cm
-1
Scaled (by 0.9891) Zero Point Vibrational Energy (zpe) 28706.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
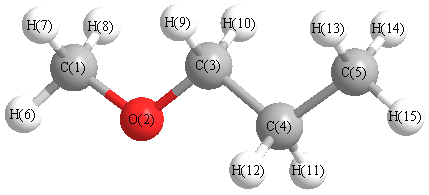
Charges from optimized geometry at LSDA/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.155 |
|
|
|
| 2 |
O |
-0.213 |
|
|
|
| 3 |
C |
-0.001 |
|
|
|
| 4 |
C |
-0.222 |
|
|
|
| 5 |
C |
-0.364 |
|
|
|
| 6 |
H |
0.117 |
|
|
|
| 7 |
H |
0.067 |
|
|
|
| 8 |
H |
0.067 |
|
|
|
| 9 |
H |
0.057 |
|
|
|
| 10 |
H |
0.057 |
|
|
|
| 11 |
H |
0.121 |
|
|
|
| 12 |
H |
0.121 |
|
|
|
| 13 |
H |
0.111 |
|
|
|
| 14 |
H |
0.111 |
|
|
|
| 15 |
H |
0.124 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.253 |
0.894 |
0.000 |
0.929 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.682 |
-2.059 |
0.000 |
| y |
-2.059 |
-34.124 |
0.000 |
| z |
0.000 |
0.000 |
-33.596 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.178 |
-2.059 |
0.000 |
| y |
-2.059 |
-1.485 |
0.000 |
| z |
0.000 |
0.000 |
-0.693 |
|
| Polar |
|---|
| 3z2-r2 | -1.386 |
|---|
| x2-y2 | 2.442 |
|---|
| xy | -2.059 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.243 |
-0.530 |
0.000 |
| y |
-0.530 |
8.040 |
0.000 |
| z |
0.000 |
0.000 |
7.661 |
<r2> (average value of r
2) Å
2
| <r2> |
176.969 |
| (<r2>)1/2 |
13.303 |