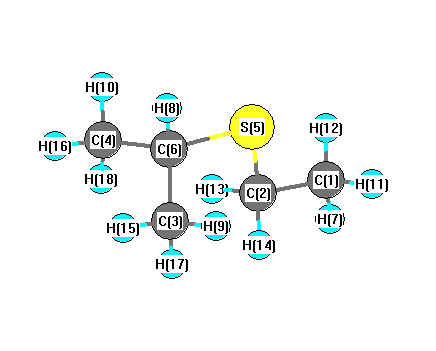
Vibrational Frequencies calculated at LSDA/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3096 |
3034 |
14.00 |
|
|
|
| 2 |
A |
3084 |
3021 |
14.31 |
|
|
|
| 3 |
A |
3061 |
2999 |
50.56 |
|
|
|
| 4 |
A |
3014 |
2953 |
2.42 |
|
|
|
| 5 |
A |
3002 |
2941 |
19.49 |
|
|
|
| 6 |
A |
2992 |
2932 |
21.26 |
|
|
|
| 7 |
A |
2978 |
2918 |
26.00 |
|
|
|
| 8 |
A |
1486 |
1456 |
12.21 |
|
|
|
| 9 |
A |
1484 |
1454 |
12.45 |
|
|
|
| 10 |
A |
1478 |
1448 |
25.78 |
|
|
|
| 11 |
A |
1463 |
1433 |
1.16 |
|
|
|
| 12 |
A |
1401 |
1373 |
25.77 |
|
|
|
| 13 |
A |
1399 |
1371 |
4.76 |
|
|
|
| 14 |
A |
1264 |
1238 |
18.17 |
|
|
|
| 15 |
A |
1260 |
1234 |
12.80 |
|
|
|
| 16 |
A |
1180 |
1156 |
8.01 |
|
|
|
| 17 |
A |
1102 |
1080 |
20.87 |
|
|
|
| 18 |
A |
1059 |
1037 |
18.23 |
|
|
|
| 19 |
A |
1014 |
993 |
10.39 |
|
|
|
| 20 |
A |
921 |
902 |
1.76 |
|
|
|
| 21 |
A |
663 |
649 |
1.33 |
|
|
|
| 22 |
A |
595 |
583 |
2.90 |
|
|
|
| 23 |
A |
457 |
448 |
3.24 |
|
|
|
| 24 |
A |
364 |
357 |
0.47 |
|
|
|
| 25 |
A |
289 |
283 |
1.96 |
|
|
|
| 26 |
A |
259 |
254 |
0.45 |
|
|
|
| 27 |
A |
159 |
156 |
0.34 |
|
|
|
| 28 |
A |
3094 |
3032 |
4.02 |
|
|
|
| 29 |
A |
3093 |
3030 |
25.81 |
|
|
|
| 30 |
A |
3058 |
2996 |
4.94 |
|
|
|
| 31 |
A |
3054 |
2992 |
0.01 |
|
|
|
| 32 |
A |
2976 |
2915 |
13.90 |
|
|
|
| 33 |
A |
1472 |
1442 |
10.43 |
|
|
|
| 34 |
A |
1466 |
1436 |
7.37 |
|
|
|
| 35 |
A |
1459 |
1429 |
0.74 |
|
|
|
| 36 |
A |
1384 |
1356 |
31.16 |
|
|
|
| 37 |
A |
1319 |
1292 |
0.71 |
|
|
|
| 38 |
A |
1242 |
1217 |
0.69 |
|
|
|
| 39 |
A |
1162 |
1138 |
1.50 |
|
|
|
| 40 |
A |
1025 |
1004 |
0.00 |
|
|
|
| 41 |
A |
958 |
939 |
0.05 |
|
|
|
| 42 |
A |
929 |
910 |
3.54 |
|
|
|
| 43 |
A |
799 |
783 |
9.46 |
|
|
|
| 44 |
A |
315 |
308 |
2.11 |
|
|
|
| 45 |
A |
249 |
244 |
0.00 |
|
|
|
| 46 |
A |
237 |
232 |
0.06 |
|
|
|
| 47 |
A |
74 |
72 |
2.01 |
|
|
|
| 48 |
A |
23 |
22 |
0.54 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 34956.0 cm
-1
Scaled (by 0.9797) Zero Point Vibrational Energy (zpe) 34246.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at LSDA/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.505 |
|
|
|
| 2 |
C |
-0.538 |
|
|
|
| 3 |
C |
-0.480 |
|
|
|
| 4 |
C |
-0.480 |
|
|
|
| 5 |
S |
0.198 |
|
|
|
| 6 |
C |
-0.380 |
|
|
|
| 7 |
H |
0.174 |
|
|
|
| 8 |
H |
0.203 |
|
|
|
| 9 |
H |
0.187 |
|
|
|
| 10 |
H |
0.187 |
|
|
|
| 11 |
H |
0.189 |
|
|
|
| 12 |
H |
0.189 |
|
|
|
| 13 |
H |
0.194 |
|
|
|
| 14 |
H |
0.194 |
|
|
|
| 15 |
H |
0.169 |
|
|
|
| 16 |
H |
0.169 |
|
|
|
| 17 |
H |
0.165 |
|
|
|
| 18 |
H |
0.165 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.025 |
-2.004 |
0.000 |
2.004 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.285 |
0.373 |
0.000 |
| y |
0.373 |
-49.621 |
0.000 |
| z |
0.000 |
0.000 |
-47.877 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.464 |
0.373 |
0.000 |
| y |
0.373 |
-4.540 |
0.000 |
| z |
0.000 |
0.000 |
-1.925 |
|
| Polar |
|---|
| 3z2-r2 | -3.849 |
|---|
| x2-y2 | 7.336 |
|---|
| xy | 0.373 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.962 |
1.113 |
0.000 |
| y |
1.113 |
10.169 |
0.000 |
| z |
0.000 |
0.000 |
9.115 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |