Jump to
S1C2
Vibrational Frequencies calculated at SVWN/6-31G*
Geometric Data calculated at SVWN/6-31G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at SVWN/6-31G*
| | hartrees |
|---|
| Energy at 0K | -622.879204 |
| Energy at 298.15K | -622.882895 |
| HF Energy | -622.879204 |
| Nuclear repulsion energy | 191.557223 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at SVWN/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3531 |
3464 |
79.35 |
|
|
|
| 2 |
A' |
1231 |
1208 |
145.78 |
|
|
|
| 3 |
A' |
1086 |
1066 |
6.11 |
|
|
|
| 4 |
A' |
764 |
749 |
129.79 |
|
|
|
| 5 |
A' |
470 |
461 |
77.96 |
|
|
|
| 6 |
A' |
426 |
418 |
97.97 |
|
|
|
| 7 |
A' |
291 |
286 |
15.66 |
|
|
|
| 8 |
A" |
3528 |
3461 |
25.52 |
|
|
|
| 9 |
A" |
1045 |
1025 |
66.52 |
|
|
|
| 10 |
A" |
774 |
759 |
332.18 |
|
|
|
| 11 |
A" |
432 |
424 |
76.87 |
|
|
|
| 12 |
A" |
165 |
162 |
13.45 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 6871.6 cm
-1
Scaled (by 0.981) Zero Point Vibrational Energy (zpe) 6741.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at SVWN/6-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.302 |
0.342 |
0.000 |
| O2 |
-1.048 |
0.946 |
0.000 |
| O3 |
0.302 |
-0.703 |
1.275 |
| O4 |
0.302 |
-0.703 |
-1.275 |
| H5 |
-0.648 |
-0.896 |
1.476 |
| H6 |
-0.648 |
-0.896 |
-1.476 |
Atom - Atom Distances (Å)
| |
S1 |
O2 |
O3 |
O4 |
H5 |
H6 |
| S1 | | 1.4787 | 1.6491 | 1.6491 | 2.1487 | 2.1487 |
O2 | 1.4787 | | 2.4836 | 2.4836 | 2.3945 | 2.3945 | O3 | 1.6491 | 2.4836 | | 2.5503 | 0.9901 | 2.9174 | O4 | 1.6491 | 2.4836 | 2.5503 | | 2.9174 | 0.9901 | H5 | 2.1487 | 2.3945 | 0.9901 | 2.9174 | | 2.9529 | H6 | 2.1487 | 2.3945 | 2.9174 | 0.9901 | 2.9529 | |
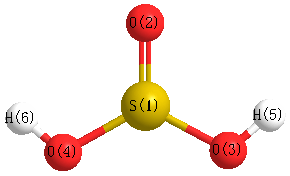 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
O3 |
H5 |
106.308 |
|
S1 |
O4 |
H6 |
106.308 |
| O2 |
S1 |
O3 |
105.001 |
|
O2 |
S1 |
O4 |
105.001 |
| O3 |
S1 |
O4 |
101.291 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at SVWN/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.890 |
|
|
|
| 2 |
O |
-0.525 |
|
|
|
| 3 |
O |
-0.600 |
|
|
|
| 4 |
O |
-0.600 |
|
|
|
| 5 |
H |
0.418 |
|
|
|
| 6 |
H |
0.418 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.197 |
-1.348 |
0.000 |
1.803 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.944 |
5.348 |
0.000 |
| y |
5.348 |
-29.985 |
0.000 |
| z |
0.000 |
0.000 |
-28.219 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.842 |
5.348 |
0.000 |
| y |
5.348 |
-0.904 |
0.000 |
| z |
0.000 |
0.000 |
1.746 |
|
| Polar |
|---|
| 3z2-r2 | 3.492 |
|---|
| x2-y2 | 0.041 |
|---|
| xy | 5.348 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.917 |
-0.260 |
0.000 |
| y |
-0.260 |
3.855 |
0.000 |
| z |
0.000 |
0.000 |
4.431 |
<r2> (average value of r
2) Å
2
| <r2> |
79.827 |
| (<r2>)1/2 |
8.935 |