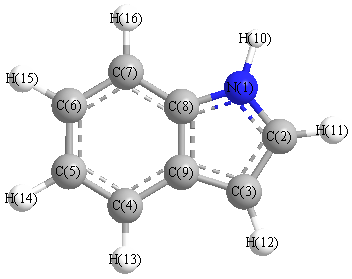
Vibrational Frequencies calculated at SVWN/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3583 |
3542 |
81.76 |
|
|
|
| 2 |
A' |
3197 |
3160 |
0.01 |
|
|
|
| 3 |
A' |
3180 |
3143 |
0.92 |
|
|
|
| 4 |
A' |
3130 |
3094 |
6.00 |
|
|
|
| 5 |
A' |
3119 |
3083 |
12.17 |
|
|
|
| 6 |
A' |
3109 |
3073 |
1.21 |
|
|
|
| 7 |
A' |
3104 |
3068 |
1.57 |
|
|
|
| 8 |
A' |
1658 |
1639 |
1.96 |
|
|
|
| 9 |
A' |
1608 |
1589 |
2.22 |
|
|
|
| 10 |
A' |
1535 |
1518 |
5.77 |
|
|
|
| 11 |
A' |
1501 |
1484 |
4.40 |
|
|
|
| 12 |
A' |
1477 |
1460 |
15.14 |
|
|
|
| 13 |
A' |
1435 |
1419 |
6.41 |
|
|
|
| 14 |
A' |
1391 |
1375 |
45.63 |
|
|
|
| 15 |
A' |
1345 |
1329 |
10.86 |
|
|
|
| 16 |
A' |
1293 |
1278 |
4.85 |
|
|
|
| 17 |
A' |
1234 |
1220 |
8.27 |
|
|
|
| 18 |
A' |
1206 |
1192 |
3.29 |
|
|
|
| 19 |
A' |
1137 |
1124 |
0.73 |
|
|
|
| 20 |
A' |
1116 |
1103 |
0.88 |
|
|
|
| 21 |
A' |
1092 |
1079 |
27.65 |
|
|
|
| 22 |
A' |
1054 |
1042 |
5.66 |
|
|
|
| 23 |
A' |
1024 |
1012 |
9.24 |
|
|
|
| 24 |
A' |
896 |
886 |
7.06 |
|
|
|
| 25 |
A' |
870 |
860 |
0.61 |
|
|
|
| 26 |
A' |
772 |
763 |
1.93 |
|
|
|
| 27 |
A' |
607 |
600 |
1.39 |
|
|
|
| 28 |
A' |
544 |
537 |
0.06 |
|
|
|
| 29 |
A' |
396 |
391 |
4.13 |
|
|
|
| 30 |
A" |
911 |
901 |
0.01 |
|
|
|
| 31 |
A" |
873 |
863 |
1.62 |
|
|
|
| 32 |
A" |
829 |
820 |
3.52 |
|
|
|
| 33 |
A" |
810 |
801 |
0.21 |
|
|
|
| 34 |
A" |
737 |
728 |
14.32 |
|
|
|
| 35 |
A" |
717 |
709 |
107.91 |
|
|
|
| 36 |
A" |
689 |
681 |
25.08 |
|
|
|
| 37 |
A" |
607 |
600 |
8.35 |
|
|
|
| 38 |
A" |
557 |
551 |
0.47 |
|
|
|
| 39 |
A" |
439 |
434 |
62.31 |
|
|
|
| 40 |
A" |
409 |
404 |
10.77 |
|
|
|
| 41 |
A" |
237 |
234 |
0.33 |
|
|
|
| 42 |
A" |
207 |
204 |
10.22 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 27817.7 cm
-1
Scaled (by 0.9885) Zero Point Vibrational Energy (zpe) 27497.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at SVWN/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.186 |
|
|
|
| 2 |
C |
-0.135 |
|
|
|
| 3 |
C |
-0.113 |
|
|
|
| 4 |
C |
-0.040 |
|
|
|
| 5 |
C |
-0.158 |
|
|
|
| 6 |
C |
-0.169 |
|
|
|
| 7 |
C |
-0.067 |
|
|
|
| 8 |
C |
-0.081 |
|
|
|
| 9 |
C |
-0.180 |
|
|
|
| 10 |
H |
0.262 |
|
|
|
| 11 |
H |
0.157 |
|
|
|
| 12 |
H |
0.151 |
|
|
|
| 13 |
H |
0.132 |
|
|
|
| 14 |
H |
0.141 |
|
|
|
| 15 |
H |
0.138 |
|
|
|
| 16 |
H |
0.147 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.917 |
2.091 |
0.000 |
2.283 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.995 |
-2.177 |
0.000 |
| y |
-2.177 |
-42.034 |
0.000 |
| z |
0.000 |
0.000 |
-58.428 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.236 |
-2.177 |
0.000 |
| y |
-2.177 |
10.677 |
0.000 |
| z |
0.000 |
0.000 |
-13.913 |
|
| Polar |
|---|
| 3z2-r2 | -27.827 |
|---|
| x2-y2 | -4.961 |
|---|
| xy | -2.177 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
19.611 |
-1.565 |
0.000 |
| y |
-1.565 |
15.660 |
0.000 |
| z |
0.000 |
0.000 |
7.619 |
<r2> (average value of r
2) Å
2
| <r2> |
279.369 |
| (<r2>)1/2 |
16.714 |