Vibrational Frequencies calculated at PBE1PBE/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3794 |
3648 |
74.41 |
|
|
|
| 2 |
A |
3587 |
3449 |
8.05 |
|
|
|
| 3 |
A |
3166 |
3044 |
17.40 |
|
|
|
| 4 |
A |
3140 |
3020 |
11.87 |
|
|
|
| 5 |
A |
3115 |
2995 |
27.73 |
|
|
|
| 6 |
A |
3110 |
2990 |
38.48 |
|
|
|
| 7 |
A |
3090 |
2971 |
19.66 |
|
|
|
| 8 |
A |
3053 |
2936 |
19.52 |
|
|
|
| 9 |
A |
2963 |
2850 |
76.09 |
|
|
|
| 10 |
A |
1833 |
1762 |
273.26 |
|
|
|
| 11 |
A |
1510 |
1452 |
0.54 |
|
|
|
| 12 |
A |
1493 |
1436 |
6.96 |
|
|
|
| 13 |
A |
1473 |
1417 |
1.04 |
|
|
|
| 14 |
A |
1449 |
1393 |
27.14 |
|
|
|
| 15 |
A |
1373 |
1320 |
32.03 |
|
|
|
| 16 |
A |
1359 |
1307 |
3.86 |
|
|
|
| 17 |
A |
1325 |
1274 |
13.29 |
|
|
|
| 18 |
A |
1317 |
1267 |
7.89 |
|
|
|
| 19 |
A |
1305 |
1254 |
1.46 |
|
|
|
| 20 |
A |
1255 |
1207 |
7.67 |
|
|
|
| 21 |
A |
1243 |
1195 |
2.09 |
|
|
|
| 22 |
A |
1218 |
1172 |
17.87 |
|
|
|
| 23 |
A |
1198 |
1152 |
16.87 |
|
|
|
| 24 |
A |
1177 |
1131 |
210.52 |
|
|
|
| 25 |
A |
1161 |
1117 |
23.60 |
|
|
|
| 26 |
A |
1105 |
1062 |
1.03 |
|
|
|
| 27 |
A |
1090 |
1048 |
4.60 |
|
|
|
| 28 |
A |
1010 |
971 |
4.03 |
|
|
|
| 29 |
A |
979 |
942 |
9.10 |
|
|
|
| 30 |
A |
945 |
909 |
3.30 |
|
|
|
| 31 |
A |
927 |
891 |
1.36 |
|
|
|
| 32 |
A |
891 |
857 |
48.01 |
|
|
|
| 33 |
A |
845 |
813 |
27.22 |
|
|
|
| 34 |
A |
789 |
759 |
1.91 |
|
|
|
| 35 |
A |
750 |
721 |
37.07 |
|
|
|
| 36 |
A |
672 |
646 |
88.07 |
|
|
|
| 37 |
A |
626 |
602 |
53.15 |
|
|
|
| 38 |
A |
591 |
569 |
16.53 |
|
|
|
| 39 |
A |
507 |
488 |
30.81 |
|
|
|
| 40 |
A |
499 |
480 |
15.17 |
|
|
|
| 41 |
A |
350 |
337 |
2.25 |
|
|
|
| 42 |
A |
258 |
248 |
2.98 |
|
|
|
| 43 |
A |
183 |
176 |
0.18 |
|
|
|
| 44 |
A |
56 |
54 |
0.26 |
|
|
|
| 45 |
A |
35 |
33 |
2.35 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31906.8 cm
-1
Scaled (by 0.9616) Zero Point Vibrational Energy (zpe) 30681.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
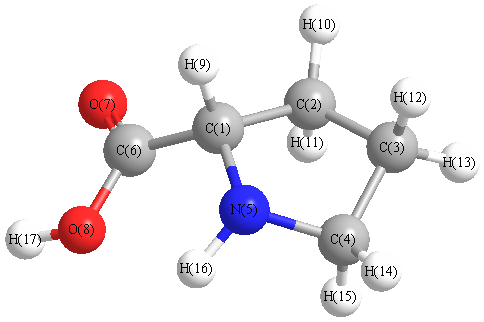
Charges from optimized geometry at PBE1PBE/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.238 |
|
|
|
| 2 |
C |
0.716 |
|
|
|
| 3 |
C |
0.651 |
|
|
|
| 4 |
C |
0.953 |
|
|
|
| 5 |
N |
-0.399 |
|
|
|
| 6 |
C |
1.028 |
|
|
|
| 7 |
O |
-0.624 |
|
|
|
| 8 |
O |
-0.581 |
|
|
|
| 9 |
H |
-0.291 |
|
|
|
| 10 |
H |
-0.230 |
|
|
|
| 11 |
H |
-0.359 |
|
|
|
| 12 |
H |
-0.292 |
|
|
|
| 13 |
H |
-0.244 |
|
|
|
| 14 |
H |
-0.345 |
|
|
|
| 15 |
H |
-0.349 |
|
|
|
| 16 |
H |
-0.053 |
|
|
|
| 17 |
H |
0.181 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.236 |
-1.353 |
-0.349 |
1.417 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.794 |
6.001 |
-1.337 |
| y |
6.001 |
-50.076 |
2.776 |
| z |
-1.337 |
2.776 |
-47.997 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.242 |
6.001 |
-1.337 |
| y |
6.001 |
-2.680 |
2.776 |
| z |
-1.337 |
2.776 |
0.438 |
|
| Polar |
|---|
| 3z2-r2 | 0.876 |
|---|
| x2-y2 | 3.282 |
|---|
| xy | 6.001 |
|---|
| xz | -1.337 |
|---|
| yz | 2.776 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.887 |
-0.017 |
0.116 |
| y |
-0.017 |
11.137 |
0.101 |
| z |
0.116 |
0.101 |
9.580 |
<r2> (average value of r
2) Å
2
| <r2> |
259.124 |
| (<r2>)1/2 |
16.097 |