Jump to
S1C2
Energy calculated at PBE1PBE/6-31G
| | hartrees |
|---|
| Energy at 0K | -750.451720 |
| Energy at 298.15K | |
| HF Energy | -750.451720 |
| Nuclear repulsion energy | 554.686896 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBE1PBE/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
1873 |
1790 |
0.00 |
|
|
|
| 2 |
Ag |
1446 |
1382 |
0.00 |
|
|
|
| 3 |
Ag |
1329 |
1270 |
0.00 |
|
|
|
| 4 |
Ag |
1186 |
1134 |
0.00 |
|
|
|
| 5 |
Ag |
698 |
667 |
0.00 |
|
|
|
| 6 |
Ag |
573 |
547 |
0.00 |
|
|
|
| 7 |
Ag |
373 |
356 |
0.00 |
|
|
|
| 8 |
Ag |
338 |
324 |
0.00 |
|
|
|
| 9 |
Ag |
217 |
208 |
0.00 |
|
|
|
| 10 |
Au |
541 |
517 |
2.70 |
|
|
|
| 11 |
Au |
328 |
313 |
9.24 |
|
|
|
| 12 |
Au |
110 |
106 |
0.24 |
|
|
|
| 13 |
Au |
16i |
16i |
0.08 |
|
|
|
| 14 |
Bg |
646 |
618 |
0.00 |
|
|
|
| 15 |
Bg |
450 |
430 |
0.00 |
|
|
|
| 16 |
Bg |
172 |
164 |
0.00 |
|
|
|
| 17 |
Bu |
1820 |
1739 |
277.82 |
|
|
|
| 18 |
Bu |
1343 |
1284 |
311.46 |
|
|
|
| 19 |
Bu |
1213 |
1159 |
185.72 |
|
|
|
| 20 |
Bu |
947 |
905 |
217.57 |
|
|
|
| 21 |
Bu |
599 |
573 |
4.77 |
|
|
|
| 22 |
Bu |
471 |
450 |
5.97 |
|
|
|
| 23 |
Bu |
284 |
271 |
6.34 |
|
|
|
| 24 |
Bu |
134 |
128 |
1.35 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8536.7 cm
-1
Scaled (by 0.9559) Zero Point Vibrational Energy (zpe) 8160.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBE1PBE/6-31G
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.154 |
1.857 |
0.000 |
| C2 |
0.458 |
0.556 |
0.000 |
| C3 |
-0.458 |
-0.556 |
0.000 |
| C4 |
-0.154 |
-1.857 |
0.000 |
| F5 |
1.097 |
2.825 |
0.000 |
| F6 |
-1.097 |
2.363 |
0.000 |
| F7 |
1.795 |
0.222 |
0.000 |
| F8 |
-1.795 |
-0.222 |
0.000 |
| F9 |
1.097 |
-2.363 |
0.000 |
| F10 |
-1.097 |
-2.825 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| C1 | | 1.3359 | 2.4898 | 3.7271 | 1.3505 | 1.3494 | 2.3162 | 2.8506 | 4.3241 | 4.8461 |
C2 | 1.3359 | | 1.4408 | 2.4898 | 2.3568 | 2.3835 | 1.3785 | 2.3839 | 2.9883 | 3.7211 | C3 | 2.4898 | 1.4408 | | 1.3359 | 3.7211 | 2.9883 | 2.3839 | 1.3785 | 2.3835 | 2.3568 | C4 | 3.7271 | 2.4898 | 1.3359 | | 4.8461 | 4.3241 | 2.8506 | 2.3162 | 1.3494 | 1.3505 | F5 | 1.3505 | 2.3568 | 3.7211 | 4.8461 | | 2.2411 | 2.6945 | 4.2010 | 5.1879 | 6.0602 | F6 | 1.3494 | 2.3835 | 2.9883 | 4.3241 | 2.2411 | | 3.5979 | 2.6783 | 5.2103 | 5.1879 | F7 | 2.3162 | 1.3785 | 2.3839 | 2.8506 | 2.6945 | 3.5979 | | 3.6181 | 2.6783 | 4.2010 | F8 | 2.8506 | 2.3839 | 1.3785 | 2.3162 | 4.2010 | 2.6783 | 3.6181 | | 3.5979 | 2.6945 | F9 | 4.3241 | 2.9883 | 2.3835 | 1.3494 | 5.1879 | 5.2103 | 2.6783 | 3.5979 | | 2.2411 | F10 | 4.8461 | 3.7211 | 2.3568 | 1.3505 | 6.0602 | 5.1879 | 4.2010 | 2.6945 | 2.2411 | |
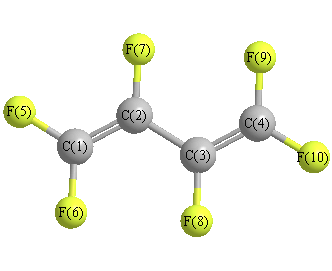 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
127.411 |
|
C1 |
C2 |
F7 |
117.138 |
| C2 |
C1 |
F5 |
122.637 |
|
C2 |
C1 |
F6 |
125.151 |
| C2 |
C3 |
C4 |
127.411 |
|
C2 |
C3 |
F8 |
115.451 |
| C3 |
C2 |
F7 |
115.451 |
|
C3 |
C4 |
F9 |
125.151 |
| C3 |
C4 |
F10 |
122.637 |
|
C4 |
C3 |
F8 |
117.138 |
| F5 |
C1 |
F6 |
112.212 |
|
F9 |
C4 |
F10 |
112.212 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBE1PBE/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.563 |
|
|
|
| 2 |
C |
0.250 |
|
|
|
| 3 |
C |
0.250 |
|
|
|
| 4 |
C |
0.563 |
|
|
|
| 5 |
F |
-0.262 |
|
|
|
| 6 |
F |
-0.258 |
|
|
|
| 7 |
F |
-0.293 |
|
|
|
| 8 |
F |
-0.293 |
|
|
|
| 9 |
F |
-0.258 |
|
|
|
| 10 |
F |
-0.262 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-57.741 |
-0.179 |
0.000 |
| y |
-0.179 |
-55.708 |
0.000 |
| z |
0.000 |
0.000 |
-48.994 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-5.390 |
-0.179 |
0.000 |
| y |
-0.179 |
-2.340 |
0.000 |
| z |
0.000 |
0.000 |
7.730 |
|
| Polar |
|---|
| 3z2-r2 | 15.460 |
|---|
| x2-y2 | -2.033 |
|---|
| xy | -0.179 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.996 |
-0.179 |
0.000 |
| y |
-0.179 |
11.711 |
0.000 |
| z |
0.000 |
0.000 |
2.551 |
<r2> (average value of r
2) Å
2
| <r2> |
428.056 |
| (<r2>)1/2 |
20.690 |
Jump to
S1C1
Energy calculated at PBE1PBE/6-31G
| | hartrees |
|---|
| Energy at 0K | -750.451411 |
| Energy at 298.15K | -750.452369 |
| HF Energy | -750.451411 |
| Nuclear repulsion energy | 559.304429 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBE1PBE/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
1869 |
1787 |
61.20 |
|
|
|
| 2 |
A |
1413 |
1351 |
22.08 |
|
|
|
| 3 |
A |
1345 |
1286 |
116.66 |
|
|
|
| 4 |
A |
1136 |
1086 |
208.27 |
|
|
|
| 5 |
A |
697 |
666 |
0.30 |
|
|
|
| 6 |
A |
661 |
632 |
1.14 |
|
|
|
| 7 |
A |
501 |
479 |
0.07 |
|
|
|
| 8 |
A |
450 |
430 |
0.67 |
|
|
|
| 9 |
A |
370 |
354 |
1.71 |
|
|
|
| 10 |
A |
253 |
242 |
0.20 |
|
|
|
| 11 |
A |
181 |
173 |
0.42 |
|
|
|
| 12 |
A |
98 |
94 |
0.51 |
|
|
|
| 13 |
A |
54 |
52 |
0.00 |
|
|
|
| 14 |
B |
1840 |
1758 |
197.12 |
|
|
|
| 15 |
B |
1339 |
1280 |
201.37 |
|
|
|
| 16 |
B |
1210 |
1157 |
65.26 |
|
|
|
| 17 |
B |
963 |
921 |
146.53 |
|
|
|
| 18 |
B |
600 |
574 |
4.71 |
|
|
|
| 19 |
B |
585 |
559 |
1.36 |
|
|
|
| 20 |
B |
536 |
513 |
6.30 |
|
|
|
| 21 |
B |
388 |
371 |
6.45 |
|
|
|
| 22 |
B |
287 |
275 |
5.37 |
|
|
|
| 23 |
B |
204 |
195 |
5.11 |
|
|
|
| 24 |
B |
107 |
102 |
0.60 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8543.9 cm
-1
Scaled (by 0.9559) Zero Point Vibrational Energy (zpe) 8167.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBE1PBE/6-31G
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.175 |
1.554 |
-0.382 |
| C2 |
0.121 |
0.710 |
0.606 |
| C3 |
-0.121 |
-0.710 |
0.606 |
| C4 |
0.175 |
-1.554 |
-0.382 |
| F5 |
0.121 |
2.867 |
-0.373 |
| F6 |
-0.790 |
1.173 |
-1.525 |
| F7 |
0.699 |
1.226 |
1.748 |
| F8 |
-0.699 |
-1.226 |
1.748 |
| F9 |
0.790 |
-1.173 |
-1.525 |
| F10 |
-0.121 |
-2.867 |
-0.373 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| C1 | | 1.3324 | 2.4709 | 3.1275 | 1.3465 | 1.3523 | 2.3257 | 3.5410 | 3.1103 | 4.4218 |
C2 | 1.3324 | | 1.4411 | 2.4709 | 2.3689 | 2.3630 | 1.3799 | 2.3928 | 2.9214 | 3.7171 | C3 | 2.4709 | 1.4411 | | 1.3324 | 3.7171 | 2.9214 | 2.3928 | 1.3799 | 2.3630 | 2.3689 | C4 | 3.1275 | 2.4709 | 1.3324 | | 4.4218 | 3.1103 | 3.5410 | 2.3257 | 1.3523 | 1.3465 | F5 | 1.3465 | 2.3689 | 3.7171 | 4.4218 | | 2.2420 | 2.7438 | 4.6824 | 4.2546 | 5.7401 | F6 | 1.3523 | 2.3630 | 2.9214 | 3.1103 | 2.2420 | | 3.5959 | 4.0590 | 2.8285 | 4.2546 | F7 | 2.3257 | 1.3799 | 2.3928 | 3.5410 | 2.7438 | 3.5959 | | 2.8220 | 4.0590 | 4.6824 | F8 | 3.5410 | 2.3928 | 1.3799 | 2.3257 | 4.6824 | 4.0590 | 2.8220 | | 3.5959 | 2.7438 | F9 | 3.1103 | 2.9214 | 2.3630 | 1.3523 | 4.2546 | 2.8285 | 4.0590 | 3.5959 | | 2.2420 | F10 | 4.4218 | 3.7171 | 2.3689 | 1.3465 | 5.7401 | 4.2546 | 4.6824 | 2.7438 | 2.2420 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
125.927 |
|
C1 |
C2 |
F7 |
118.051 |
| C2 |
C1 |
F5 |
124.322 |
|
C2 |
C1 |
F6 |
123.321 |
| C2 |
C3 |
C4 |
125.927 |
|
C2 |
C3 |
F8 |
116.018 |
| C3 |
C2 |
F7 |
116.018 |
|
C3 |
C4 |
F9 |
123.321 |
| C3 |
C4 |
F10 |
124.322 |
|
C4 |
C3 |
F8 |
118.051 |
| F5 |
C1 |
F6 |
112.346 |
|
F9 |
C4 |
F10 |
112.346 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBE1PBE/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.569 |
|
|
|
| 2 |
C |
0.242 |
|
|
|
| 3 |
C |
0.242 |
|
|
|
| 4 |
C |
0.569 |
|
|
|
| 5 |
F |
-0.257 |
|
|
|
| 6 |
F |
-0.265 |
|
|
|
| 7 |
F |
-0.288 |
|
|
|
| 8 |
F |
-0.288 |
|
|
|
| 9 |
F |
-0.265 |
|
|
|
| 10 |
F |
-0.257 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.705 |
0.705 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-50.626 |
-1.512 |
0.000 |
| y |
-1.512 |
-55.533 |
0.000 |
| z |
0.000 |
0.000 |
-56.556 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.419 |
-1.512 |
0.000 |
| y |
-1.512 |
-1.942 |
0.000 |
| z |
0.000 |
0.000 |
-3.477 |
|
| Polar |
|---|
| 3z2-r2 | -6.953 |
|---|
| x2-y2 | 4.907 |
|---|
| xy | -1.512 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.399 |
-0.034 |
0.000 |
| y |
-0.034 |
8.837 |
0.000 |
| z |
0.000 |
0.000 |
6.650 |
<r2> (average value of r
2) Å
2
| <r2> |
395.074 |
| (<r2>)1/2 |
19.876 |