Vibrational Frequencies calculated at B1B95/CEP-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3774 |
3612 |
42.82 |
|
|
|
| 2 |
A |
3634 |
3478 |
57.21 |
|
|
|
| 3 |
A |
3273 |
3133 |
6.73 |
|
|
|
| 4 |
A |
3193 |
3056 |
15.23 |
|
|
|
| 5 |
A |
3171 |
3036 |
9.51 |
|
|
|
| 6 |
A |
1808 |
1731 |
302.92 |
|
|
|
| 7 |
A |
1719 |
1646 |
32.07 |
|
|
|
| 8 |
A |
1616 |
1546 |
143.98 |
|
|
|
| 9 |
A |
1423 |
1362 |
118.41 |
|
|
|
| 10 |
A |
1351 |
1294 |
44.34 |
|
|
|
| 11 |
A |
1269 |
1215 |
64.05 |
|
|
|
| 12 |
A |
1096 |
1049 |
0.72 |
|
|
|
| 13 |
A |
1022 |
978 |
47.95 |
|
|
|
| 14 |
A |
1009 |
966 |
4.33 |
|
|
|
| 15 |
A |
1000 |
958 |
33.30 |
|
|
|
| 16 |
A |
821 |
786 |
17.42 |
|
|
|
| 17 |
A |
819 |
784 |
8.42 |
|
|
|
| 18 |
A |
612 |
586 |
6.73 |
|
|
|
| 19 |
A |
596 |
570 |
5.77 |
|
|
|
| 20 |
A |
473 |
452 |
14.71 |
|
|
|
| 21 |
A |
446 |
427 |
4.56 |
|
|
|
| 22 |
A |
250 |
239 |
7.29 |
|
|
|
| 23 |
A |
164 |
157 |
188.48 |
|
|
|
| 24 |
A |
116 |
111 |
71.05 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17326.5 cm
-1
Scaled (by 0.9572) Zero Point Vibrational Energy (zpe) 16584.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
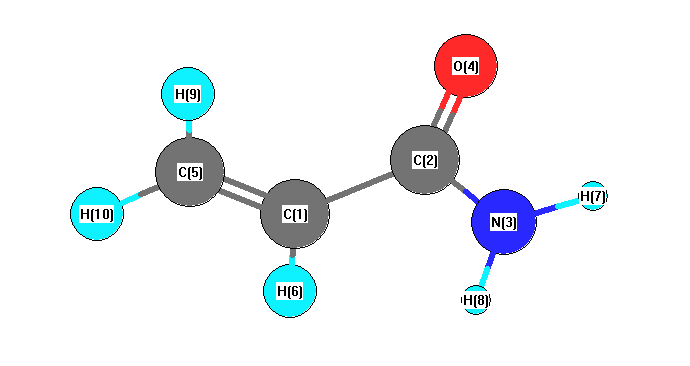
Charges from optimized geometry at B1B95/CEP-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.042 |
|
|
|
| 2 |
C |
-0.335 |
|
|
|
| 3 |
N |
-0.368 |
|
|
|
| 4 |
O |
-0.114 |
|
|
|
| 5 |
C |
-0.647 |
|
|
|
| 6 |
H |
0.288 |
|
|
|
| 7 |
H |
0.336 |
|
|
|
| 8 |
H |
0.325 |
|
|
|
| 9 |
H |
0.266 |
|
|
|
| 10 |
H |
0.208 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.428 |
-3.743 |
0.316 |
3.780 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-20.889 |
3.131 |
-0.681 |
| y |
3.131 |
-29.489 |
-0.326 |
| z |
-0.681 |
-0.326 |
-31.803 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
9.758 |
3.131 |
-0.681 |
| y |
3.131 |
-3.143 |
-0.326 |
| z |
-0.681 |
-0.326 |
-6.615 |
|
| Polar |
|---|
| 3z2-r2 | -13.229 |
|---|
| x2-y2 | 8.600 |
|---|
| xy | 3.131 |
|---|
| xz | -0.681 |
|---|
| yz | -0.326 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.779 |
0.892 |
0.019 |
| y |
0.892 |
6.406 |
0.020 |
| z |
0.019 |
0.020 |
3.204 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |