Jump to
S1C2
Energy calculated at B1B95/6-311G**
| | hartrees |
|---|
| Energy at 0K | -312.190237 |
| Energy at 298.15K | -312.205719 |
| HF Energy | -312.190237 |
| Nuclear repulsion energy | 316.141699 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3136 |
3011 |
46.34 |
|
|
|
| 2 |
A1 |
3068 |
2945 |
52.37 |
|
|
|
| 3 |
A1 |
3053 |
2931 |
17.97 |
|
|
|
| 4 |
A1 |
2976 |
2857 |
97.55 |
|
|
|
| 5 |
A1 |
1536 |
1475 |
2.76 |
|
|
|
| 6 |
A1 |
1510 |
1450 |
8.14 |
|
|
|
| 7 |
A1 |
1496 |
1436 |
0.00 |
|
|
|
| 8 |
A1 |
1458 |
1400 |
0.29 |
|
|
|
| 9 |
A1 |
1413 |
1356 |
2.24 |
|
|
|
| 10 |
A1 |
1338 |
1285 |
2.20 |
|
|
|
| 11 |
A1 |
1184 |
1137 |
3.08 |
|
|
|
| 12 |
A1 |
1083 |
1040 |
9.20 |
|
|
|
| 13 |
A1 |
1028 |
987 |
17.76 |
|
|
|
| 14 |
A1 |
927 |
890 |
8.36 |
|
|
|
| 15 |
A1 |
425 |
408 |
0.54 |
|
|
|
| 16 |
A1 |
318 |
305 |
0.27 |
|
|
|
| 17 |
A1 |
101 |
97 |
0.11 |
|
|
|
| 18 |
A2 |
3128 |
3003 |
0.00 |
|
|
|
| 19 |
A2 |
3102 |
2978 |
0.00 |
|
|
|
| 20 |
A2 |
3001 |
2881 |
0.00 |
|
|
|
| 21 |
A2 |
1500 |
1440 |
0.00 |
|
|
|
| 22 |
A2 |
1316 |
1263 |
0.00 |
|
|
|
| 23 |
A2 |
1267 |
1217 |
0.00 |
|
|
|
| 24 |
A2 |
1179 |
1132 |
0.00 |
|
|
|
| 25 |
A2 |
895 |
859 |
0.00 |
|
|
|
| 26 |
A2 |
766 |
735 |
0.00 |
|
|
|
| 27 |
A2 |
228 |
219 |
0.00 |
|
|
|
| 28 |
A2 |
135 |
129 |
0.00 |
|
|
|
| 29 |
A2 |
74 |
71 |
0.00 |
|
|
|
| 30 |
B1 |
3128 |
3003 |
130.40 |
|
|
|
| 31 |
B1 |
3102 |
2978 |
3.07 |
|
|
|
| 32 |
B1 |
2997 |
2878 |
116.75 |
|
|
|
| 33 |
B1 |
1500 |
1440 |
16.80 |
|
|
|
| 34 |
B1 |
1318 |
1265 |
1.11 |
|
|
|
| 35 |
B1 |
1275 |
1224 |
4.01 |
|
|
|
| 36 |
B1 |
1201 |
1153 |
4.62 |
|
|
|
| 37 |
B1 |
909 |
872 |
3.60 |
|
|
|
| 38 |
B1 |
768 |
737 |
3.86 |
|
|
|
| 39 |
B1 |
230 |
221 |
0.23 |
|
|
|
| 40 |
B1 |
156 |
150 |
5.32 |
|
|
|
| 41 |
B1 |
55 |
53 |
0.22 |
|
|
|
| 42 |
B2 |
3136 |
3011 |
22.60 |
|
|
|
| 43 |
B2 |
3067 |
2945 |
3.49 |
|
|
|
| 44 |
B2 |
3052 |
2931 |
40.10 |
|
|
|
| 45 |
B2 |
2965 |
2847 |
13.34 |
|
|
|
| 46 |
B2 |
1519 |
1458 |
10.57 |
|
|
|
| 47 |
B2 |
1507 |
1447 |
0.68 |
|
|
|
| 48 |
B2 |
1494 |
1435 |
1.81 |
|
|
|
| 49 |
B2 |
1414 |
1357 |
2.71 |
|
|
|
| 50 |
B2 |
1409 |
1352 |
40.54 |
|
|
|
| 51 |
B2 |
1323 |
1270 |
9.55 |
|
|
|
| 52 |
B2 |
1190 |
1143 |
254.44 |
|
|
|
| 53 |
B2 |
1133 |
1088 |
2.70 |
|
|
|
| 54 |
B2 |
1070 |
1027 |
0.18 |
|
|
|
| 55 |
B2 |
906 |
870 |
2.05 |
|
|
|
| 56 |
B2 |
513 |
493 |
5.78 |
|
|
|
| 57 |
B2 |
243 |
233 |
0.69 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 42608.4 cm
-1
Scaled (by 0.9601) Zero Point Vibrational Energy (zpe) 40908.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/6-311G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.386 |
| C2 |
0.000 |
1.171 |
-0.390 |
| C3 |
0.000 |
-1.171 |
-0.390 |
| C4 |
0.000 |
2.370 |
0.531 |
| C5 |
0.000 |
-2.370 |
0.531 |
| C6 |
0.000 |
3.685 |
-0.230 |
| C7 |
0.000 |
-3.685 |
-0.230 |
| H8 |
0.885 |
1.193 |
-1.044 |
| H9 |
-0.885 |
1.193 |
-1.044 |
| H10 |
-0.885 |
-1.193 |
-1.044 |
| H11 |
0.885 |
-1.193 |
-1.044 |
| H12 |
0.876 |
2.303 |
1.180 |
| H13 |
-0.876 |
2.303 |
1.180 |
| H14 |
-0.876 |
-2.303 |
1.180 |
| H15 |
0.876 |
-2.303 |
1.180 |
| H16 |
0.000 |
4.535 |
0.452 |
| H17 |
-0.881 |
3.775 |
-0.869 |
| H18 |
0.881 |
3.775 |
-0.869 |
| H19 |
0.000 |
-4.535 |
0.452 |
| H20 |
0.881 |
-3.775 |
-0.869 |
| H21 |
-0.881 |
-3.775 |
-0.869 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4050 | 1.4050 | 2.3739 | 2.3739 | 3.7361 | 3.7361 | 2.0620 | 2.0620 | 2.0620 | 2.0620 | 2.5888 | 2.5888 | 2.5888 | 2.5888 | 4.5352 | 4.0748 | 4.0748 | 4.5352 | 4.0748 | 4.0748 |
C2 | 1.4050 | | 2.3422 | 1.5114 | 3.6584 | 2.5189 | 4.8586 | 1.1004 | 1.1004 | 2.6077 | 2.6077 | 2.1244 | 2.1244 | 3.9119 | 3.9119 | 3.4674 | 2.7906 | 2.7906 | 5.7676 | 5.0470 | 5.0470 | C3 | 1.4050 | 2.3422 | | 3.6584 | 1.5114 | 4.8586 | 2.5189 | 2.6077 | 2.6077 | 1.1004 | 1.1004 | 3.9119 | 3.9119 | 2.1244 | 2.1244 | 5.7676 | 5.0470 | 5.0470 | 3.4674 | 2.7906 | 2.7906 | C4 | 2.3739 | 1.5114 | 3.6584 | | 4.7391 | 1.5197 | 6.1021 | 2.1556 | 2.1556 | 3.9944 | 3.9944 | 1.0920 | 1.0920 | 4.7983 | 4.7983 | 2.1667 | 2.1705 | 2.1705 | 6.9047 | 6.3635 | 6.3635 | C5 | 2.3739 | 3.6584 | 1.5114 | 4.7391 | | 6.1021 | 1.5197 | 3.9944 | 3.9944 | 2.1556 | 2.1556 | 4.7983 | 4.7983 | 1.0920 | 1.0920 | 6.9047 | 6.3635 | 6.3635 | 2.1667 | 2.1705 | 2.1705 | C6 | 3.7361 | 2.5189 | 4.8586 | 1.5197 | 6.1021 | | 7.3698 | 2.7665 | 2.7665 | 5.0240 | 5.0240 | 2.1595 | 2.1595 | 6.2140 | 6.2140 | 1.0898 | 1.0918 | 1.0918 | 8.2479 | 7.5391 | 7.5391 | C7 | 3.7361 | 4.8586 | 2.5189 | 6.1021 | 1.5197 | 7.3698 | | 5.0240 | 5.0240 | 2.7665 | 2.7665 | 6.2140 | 6.2140 | 2.1595 | 2.1595 | 8.2479 | 7.5391 | 7.5391 | 1.0898 | 1.0918 | 1.0918 | H8 | 2.0620 | 1.1004 | 2.6077 | 2.1556 | 3.9944 | 2.7665 | 5.0240 | | 1.7696 | 2.9709 | 2.3864 | 2.4854 | 3.0457 | 4.5022 | 4.1437 | 3.7665 | 3.1330 | 2.5881 | 5.9858 | 4.9716 | 5.2758 | H9 | 2.0620 | 1.1004 | 2.6077 | 2.1556 | 3.9944 | 2.7665 | 5.0240 | 1.7696 | | 2.3864 | 2.9709 | 3.0457 | 2.4854 | 4.1437 | 4.5022 | 3.7665 | 2.5881 | 3.1330 | 5.9858 | 5.2758 | 4.9716 | H10 | 2.0620 | 2.6077 | 1.1004 | 3.9944 | 2.1556 | 5.0240 | 2.7665 | 2.9709 | 2.3864 | | 1.7696 | 4.5022 | 4.1437 | 2.4854 | 3.0457 | 5.9858 | 4.9716 | 5.2758 | 3.7665 | 3.1330 | 2.5881 | H11 | 2.0620 | 2.6077 | 1.1004 | 3.9944 | 2.1556 | 5.0240 | 2.7665 | 2.3864 | 2.9709 | 1.7696 | | 4.1437 | 4.5022 | 3.0457 | 2.4854 | 5.9858 | 5.2758 | 4.9716 | 3.7665 | 2.5881 | 3.1330 | H12 | 2.5888 | 2.1244 | 3.9119 | 1.0920 | 4.7983 | 2.1595 | 6.2140 | 2.4854 | 3.0457 | 4.5022 | 4.1437 | | 1.7515 | 4.9284 | 4.6067 | 2.5051 | 3.0739 | 2.5225 | 6.9323 | 6.4145 | 6.6507 | H13 | 2.5888 | 2.1244 | 3.9119 | 1.0920 | 4.7983 | 2.1595 | 6.2140 | 3.0457 | 2.4854 | 4.1437 | 4.5022 | 1.7515 | | 4.6067 | 4.9284 | 2.5051 | 2.5225 | 3.0739 | 6.9323 | 6.6507 | 6.4145 | H14 | 2.5888 | 3.9119 | 2.1244 | 4.7983 | 1.0920 | 6.2140 | 2.1595 | 4.5022 | 4.1437 | 2.4854 | 3.0457 | 4.9284 | 4.6067 | | 1.7515 | 6.9323 | 6.4145 | 6.6507 | 2.5051 | 3.0739 | 2.5225 | H15 | 2.5888 | 3.9119 | 2.1244 | 4.7983 | 1.0920 | 6.2140 | 2.1595 | 4.1437 | 4.5022 | 3.0457 | 2.4854 | 4.6067 | 4.9284 | 1.7515 | | 6.9323 | 6.6507 | 6.4145 | 2.5051 | 2.5225 | 3.0739 | H16 | 4.5352 | 3.4674 | 5.7676 | 2.1667 | 6.9047 | 1.0898 | 8.2479 | 3.7665 | 3.7665 | 5.9858 | 5.9858 | 2.5051 | 2.5051 | 6.9323 | 6.9323 | | 1.7599 | 1.7599 | 9.0695 | 8.4603 | 8.4603 | H17 | 4.0748 | 2.7906 | 5.0470 | 2.1705 | 6.3635 | 1.0918 | 7.5391 | 3.1330 | 2.5881 | 4.9716 | 5.2758 | 3.0739 | 2.5225 | 6.4145 | 6.6507 | 1.7599 | | 1.7619 | 8.4603 | 7.7534 | 7.5506 | H18 | 4.0748 | 2.7906 | 5.0470 | 2.1705 | 6.3635 | 1.0918 | 7.5391 | 2.5881 | 3.1330 | 5.2758 | 4.9716 | 2.5225 | 3.0739 | 6.6507 | 6.4145 | 1.7599 | 1.7619 | | 8.4603 | 7.5506 | 7.7534 | H19 | 4.5352 | 5.7676 | 3.4674 | 6.9047 | 2.1667 | 8.2479 | 1.0898 | 5.9858 | 5.9858 | 3.7665 | 3.7665 | 6.9323 | 6.9323 | 2.5051 | 2.5051 | 9.0695 | 8.4603 | 8.4603 | | 1.7599 | 1.7599 | H20 | 4.0748 | 5.0470 | 2.7906 | 6.3635 | 2.1705 | 7.5391 | 1.0918 | 4.9716 | 5.2758 | 3.1330 | 2.5881 | 6.4145 | 6.6507 | 3.0739 | 2.5225 | 8.4603 | 7.7534 | 7.5506 | 1.7599 | | 1.7619 | H21 | 4.0748 | 5.0470 | 2.7906 | 6.3635 | 2.1705 | 7.5391 | 1.0918 | 5.2758 | 4.9716 | 2.5881 | 3.1330 | 6.6507 | 6.4145 | 2.5225 | 3.0739 | 8.4603 | 7.5506 | 7.7534 | 1.7599 | 1.7619 | |
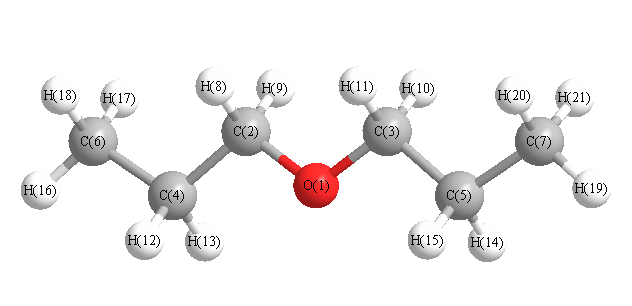 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
108.925 |
|
O1 |
C2 |
H8 |
110.184 |
| O1 |
C2 |
H9 |
110.184 |
|
O1 |
C3 |
C5 |
108.925 |
| O1 |
C3 |
H10 |
110.184 |
|
O1 |
C3 |
H11 |
110.184 |
| C2 |
O1 |
C3 |
112.925 |
|
C2 |
C4 |
C6 |
112.408 |
| C2 |
C4 |
H12 |
108.301 |
|
C2 |
C4 |
H13 |
108.301 |
| C3 |
C5 |
C7 |
112.408 |
|
C3 |
C5 |
H14 |
108.301 |
| C3 |
C5 |
H15 |
108.301 |
|
C4 |
C2 |
H8 |
110.251 |
| C4 |
C2 |
H9 |
110.251 |
|
C4 |
C6 |
H16 |
111.189 |
| C4 |
C6 |
H17 |
111.382 |
|
C4 |
C6 |
H18 |
111.382 |
| C5 |
C3 |
H10 |
110.251 |
|
C5 |
C3 |
H11 |
110.251 |
| C5 |
C7 |
H19 |
111.189 |
|
C5 |
C7 |
H20 |
111.382 |
| C5 |
C7 |
H21 |
111.382 |
|
C6 |
C4 |
H12 |
110.492 |
| C6 |
C4 |
H13 |
110.492 |
|
C7 |
C5 |
H14 |
110.492 |
| C7 |
C5 |
H15 |
110.492 |
|
H8 |
C2 |
H9 |
107.035 |
| H10 |
C3 |
H11 |
107.035 |
|
H12 |
C4 |
H13 |
106.642 |
| H14 |
C5 |
H15 |
106.642 |
|
H16 |
C6 |
H17 |
107.550 |
| H16 |
C6 |
H18 |
107.550 |
|
H17 |
C6 |
H18 |
107.589 |
| H19 |
C7 |
H20 |
107.550 |
|
H19 |
C7 |
H21 |
107.550 |
| H20 |
C7 |
H21 |
107.589 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.363 |
|
|
|
| 2 |
C |
-0.011 |
|
|
|
| 3 |
C |
-0.011 |
|
|
|
| 4 |
C |
-0.293 |
|
|
|
| 5 |
C |
-0.293 |
|
|
|
| 6 |
C |
-0.350 |
|
|
|
| 7 |
C |
-0.350 |
|
|
|
| 8 |
H |
0.099 |
|
|
|
| 9 |
H |
0.099 |
|
|
|
| 10 |
H |
0.099 |
|
|
|
| 11 |
H |
0.099 |
|
|
|
| 12 |
H |
0.135 |
|
|
|
| 13 |
H |
0.135 |
|
|
|
| 14 |
H |
0.135 |
|
|
|
| 15 |
H |
0.135 |
|
|
|
| 16 |
H |
0.128 |
|
|
|
| 17 |
H |
0.120 |
|
|
|
| 18 |
H |
0.120 |
|
|
|
| 19 |
H |
0.128 |
|
|
|
| 20 |
H |
0.120 |
|
|
|
| 21 |
H |
0.120 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.932 |
0.932 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-45.966 |
0.000 |
0.000 |
| y |
0.000 |
-43.837 |
0.000 |
| z |
0.000 |
0.000 |
-47.289 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.403 |
0.000 |
0.000 |
| y |
0.000 |
2.790 |
0.000 |
| z |
0.000 |
0.000 |
-2.387 |
|
| Polar |
|---|
| 3z2-r2 | -4.775 |
|---|
| x2-y2 | -2.128 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.660 |
0.000 |
0.000 |
| y |
0.000 |
13.833 |
0.000 |
| z |
0.000 |
0.000 |
10.127 |
<r2> (average value of r
2) Å
2
| <r2> |
430.065 |
| (<r2>)1/2 |
20.738 |
Jump to
S1C1
Energy calculated at B1B95/6-311G**
| | hartrees |
|---|
| Energy at 0K | -312.191477 |
| Energy at 298.15K | -312.207201 |
| HF Energy | -312.191477 |
| Nuclear repulsion energy | 326.992359 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3151 |
3026 |
6.06 |
|
|
|
| 2 |
A |
3129 |
3004 |
63.70 |
|
|
|
| 3 |
A |
3099 |
2975 |
10.43 |
|
|
|
| 4 |
A |
3061 |
2939 |
3.70 |
|
|
|
| 5 |
A |
3057 |
2935 |
7.05 |
|
|
|
| 6 |
A |
3010 |
2890 |
10.69 |
|
|
|
| 7 |
A |
2977 |
2859 |
100.20 |
|
|
|
| 8 |
A |
1531 |
1470 |
4.03 |
|
|
|
| 9 |
A |
1512 |
1451 |
4.24 |
|
|
|
| 10 |
A |
1494 |
1435 |
7.38 |
|
|
|
| 11 |
A |
1477 |
1418 |
1.12 |
|
|
|
| 12 |
A |
1450 |
1392 |
2.49 |
|
|
|
| 13 |
A |
1410 |
1354 |
2.86 |
|
|
|
| 14 |
A |
1376 |
1321 |
0.00 |
|
|
|
| 15 |
A |
1317 |
1265 |
3.44 |
|
|
|
| 16 |
A |
1273 |
1222 |
0.22 |
|
|
|
| 17 |
A |
1184 |
1137 |
8.29 |
|
|
|
| 18 |
A |
1148 |
1103 |
10.60 |
|
|
|
| 19 |
A |
1100 |
1056 |
8.04 |
|
|
|
| 20 |
A |
965 |
926 |
7.36 |
|
|
|
| 21 |
A |
923 |
886 |
6.87 |
|
|
|
| 22 |
A |
912 |
876 |
0.02 |
|
|
|
| 23 |
A |
752 |
722 |
0.29 |
|
|
|
| 24 |
A |
478 |
459 |
0.11 |
|
|
|
| 25 |
A |
329 |
315 |
0.32 |
|
|
|
| 26 |
A |
264 |
254 |
0.56 |
|
|
|
| 27 |
A |
166 |
159 |
0.03 |
|
|
|
| 28 |
A |
113 |
108 |
0.01 |
|
|
|
| 29 |
A |
47 |
45 |
0.00 |
|
|
|
| 30 |
B |
3151 |
3025 |
51.99 |
|
|
|
| 31 |
B |
3129 |
3004 |
30.38 |
|
|
|
| 32 |
B |
3099 |
2975 |
28.69 |
|
|
|
| 33 |
B |
3061 |
2939 |
77.29 |
|
|
|
| 34 |
B |
3057 |
2935 |
48.26 |
|
|
|
| 35 |
B |
3006 |
2886 |
103.69 |
|
|
|
| 36 |
B |
2968 |
2849 |
16.97 |
|
|
|
| 37 |
B |
1511 |
1451 |
0.71 |
|
|
|
| 38 |
B |
1509 |
1449 |
15.55 |
|
|
|
| 39 |
B |
1493 |
1434 |
9.14 |
|
|
|
| 40 |
B |
1477 |
1419 |
5.29 |
|
|
|
| 41 |
B |
1411 |
1354 |
15.70 |
|
|
|
| 42 |
B |
1397 |
1341 |
30.69 |
|
|
|
| 43 |
B |
1371 |
1316 |
13.54 |
|
|
|
| 44 |
B |
1307 |
1255 |
3.97 |
|
|
|
| 45 |
B |
1280 |
1229 |
7.44 |
|
|
|
| 46 |
B |
1193 |
1146 |
134.29 |
|
|
|
| 47 |
B |
1178 |
1131 |
63.28 |
|
|
|
| 48 |
B |
1116 |
1071 |
13.37 |
|
|
|
| 49 |
B |
1035 |
994 |
34.98 |
|
|
|
| 50 |
B |
934 |
897 |
1.00 |
|
|
|
| 51 |
B |
895 |
860 |
0.57 |
|
|
|
| 52 |
B |
785 |
754 |
2.85 |
|
|
|
| 53 |
B |
497 |
477 |
1.55 |
|
|
|
| 54 |
B |
317 |
305 |
0.48 |
|
|
|
| 55 |
B |
217 |
208 |
2.81 |
|
|
|
| 56 |
B |
171 |
164 |
4.12 |
|
|
|
| 57 |
B |
78 |
75 |
1.40 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 42671.0 cm
-1
Scaled (by 0.9601) Zero Point Vibrational Energy (zpe) 40968.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/6-311G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.112 |
| C2 |
0.028 |
1.172 |
0.888 |
| C3 |
-0.028 |
-1.172 |
0.888 |
| C4 |
0.000 |
2.371 |
-0.034 |
| C5 |
0.000 |
-2.371 |
-0.034 |
| C6 |
-1.262 |
2.434 |
-0.879 |
| C7 |
1.262 |
-2.434 |
-0.879 |
| H8 |
-0.840 |
1.194 |
1.565 |
| H9 |
0.931 |
1.187 |
1.514 |
| H10 |
0.840 |
-1.194 |
1.565 |
| H11 |
-0.931 |
-1.187 |
1.514 |
| H12 |
0.881 |
2.329 |
-0.678 |
| H13 |
0.097 |
3.273 |
0.576 |
| H14 |
-0.881 |
-2.329 |
-0.678 |
| H15 |
-0.097 |
-3.273 |
0.576 |
| H16 |
-1.244 |
3.287 |
-1.557 |
| H17 |
-1.366 |
1.527 |
-1.471 |
| H18 |
-2.150 |
2.526 |
-0.250 |
| H19 |
1.244 |
-3.287 |
-1.557 |
| H20 |
1.366 |
-1.527 |
-1.471 |
| H21 |
2.150 |
-2.526 |
-0.250 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4063 | 1.4063 | 2.3757 | 2.3757 | 2.9151 | 2.9151 | 2.0597 | 2.0593 | 2.0597 | 2.0593 | 2.6127 | 3.3076 | 2.6127 | 3.3076 | 3.8908 | 2.5888 | 3.3368 | 3.8908 | 2.5888 | 3.3368 |
C2 | 1.4063 | | 2.3452 | 1.5124 | 3.6615 | 2.5250 | 4.2012 | 1.1008 | 1.0990 | 2.5914 | 2.6220 | 2.1260 | 2.1253 | 3.9421 | 4.4584 | 3.4740 | 2.7629 | 2.8053 | 5.2293 | 3.8264 | 4.4133 | C3 | 1.4063 | 2.3452 | | 3.6615 | 1.5124 | 4.2012 | 2.5250 | 2.5914 | 2.6220 | 1.1008 | 1.0990 | 3.9421 | 4.4584 | 2.1260 | 2.1253 | 5.2293 | 3.8264 | 4.4133 | 3.4740 | 2.7629 | 2.8053 | C4 | 2.3757 | 1.5124 | 3.6615 | | 4.7425 | 1.5199 | 5.0396 | 2.1559 | 2.1597 | 3.9961 | 3.9899 | 1.0925 | 1.0931 | 4.8257 | 5.6783 | 2.1698 | 2.1554 | 2.1663 | 5.9906 | 4.3733 | 5.3529 | C5 | 2.3757 | 3.6615 | 1.5124 | 4.7425 | | 5.0396 | 1.5199 | 3.9961 | 3.9899 | 2.1559 | 2.1597 | 4.8257 | 5.6783 | 1.0925 | 1.0931 | 5.9906 | 4.3733 | 5.3529 | 2.1698 | 2.1554 | 2.1663 | C6 | 2.9151 | 2.5250 | 4.2012 | 1.5199 | 5.0396 | | 5.4835 | 2.7728 | 3.4767 | 4.8527 | 4.3523 | 2.1548 | 2.1602 | 4.7829 | 6.0040 | 1.0903 | 1.0890 | 1.0922 | 6.2828 | 4.7899 | 6.0531 | C7 | 2.9151 | 4.2012 | 2.5250 | 5.0396 | 1.5199 | 5.4835 | | 4.8527 | 4.3523 | 2.7728 | 3.4767 | 4.7829 | 6.0040 | 2.1548 | 2.1602 | 6.2828 | 4.7899 | 6.0531 | 1.0903 | 1.0890 | 1.0922 | H8 | 2.0597 | 1.1008 | 2.5914 | 2.1559 | 3.9961 | 2.7728 | 4.8527 | | 1.7716 | 2.9189 | 2.3825 | 3.0470 | 2.4864 | 4.1767 | 4.6351 | 3.7810 | 3.0996 | 2.6049 | 5.8454 | 4.6351 | 5.1058 | H9 | 2.0593 | 1.0990 | 2.6220 | 2.1597 | 3.9899 | 3.4767 | 4.3523 | 1.7716 | | 2.3825 | 3.0164 | 2.4725 | 2.4351 | 4.5222 | 4.6721 | 4.3097 | 3.7818 | 3.7943 | 5.4355 | 4.0574 | 4.2873 | H10 | 2.0597 | 2.5914 | 1.1008 | 3.9961 | 2.1559 | 4.8527 | 2.7728 | 2.9189 | 2.3825 | | 1.7716 | 4.1767 | 4.6351 | 3.0470 | 2.4864 | 5.8454 | 4.6351 | 5.1058 | 3.7810 | 3.0996 | 2.6049 | H11 | 2.0593 | 2.6220 | 1.0990 | 3.9899 | 2.1597 | 4.3523 | 3.4767 | 2.3825 | 3.0164 | 1.7716 | | 4.5222 | 4.6721 | 2.4725 | 2.4351 | 5.4355 | 4.0574 | 4.2873 | 4.3097 | 3.7818 | 3.7943 | H12 | 2.6127 | 2.1260 | 3.9421 | 1.0925 | 4.8257 | 2.1548 | 4.7829 | 3.0470 | 2.4725 | 4.1767 | 4.5222 | | 1.7546 | 4.9809 | 5.8241 | 2.4912 | 2.5144 | 3.0676 | 5.6966 | 3.9665 | 5.0369 | H13 | 3.3076 | 2.1253 | 4.4584 | 1.0931 | 5.6783 | 2.1602 | 6.0040 | 2.4864 | 2.4351 | 4.6351 | 4.6721 | 1.7546 | | 5.8241 | 6.5497 | 2.5197 | 3.0631 | 2.5079 | 6.9934 | 5.3704 | 6.2073 | H14 | 2.6127 | 3.9421 | 2.1260 | 4.8257 | 1.0925 | 4.7829 | 2.1548 | 4.1767 | 4.5222 | 3.0470 | 2.4725 | 4.9809 | 5.8241 | | 1.7546 | 5.6966 | 3.9665 | 5.0369 | 2.4912 | 2.5144 | 3.0676 | H15 | 3.3076 | 4.4584 | 2.1253 | 5.6783 | 1.0931 | 6.0040 | 2.1602 | 4.6351 | 4.6721 | 2.4864 | 2.4351 | 5.8241 | 6.5497 | 1.7546 | | 6.9934 | 5.3704 | 6.2073 | 2.5197 | 3.0631 | 2.5079 | H16 | 3.8908 | 3.4740 | 5.2293 | 2.1698 | 5.9906 | 1.0903 | 6.2828 | 3.7810 | 4.3097 | 5.8454 | 5.4355 | 2.4912 | 2.5197 | 5.6966 | 6.9934 | | 1.7669 | 1.7636 | 7.0294 | 5.4764 | 6.8574 | H17 | 2.5888 | 2.7629 | 3.8264 | 2.1554 | 4.3733 | 1.0890 | 4.7899 | 3.0996 | 3.7818 | 4.6351 | 4.0574 | 2.5144 | 3.0631 | 3.9665 | 5.3704 | 1.7669 | | 1.7626 | 5.4764 | 4.0967 | 5.5025 | H18 | 3.3368 | 2.8053 | 4.4133 | 2.1663 | 5.3529 | 1.0922 | 6.0531 | 2.6049 | 3.7943 | 5.1058 | 4.2873 | 3.0676 | 2.5079 | 5.0369 | 6.2073 | 1.7636 | 1.7626 | | 6.8574 | 5.5025 | 6.6344 | H19 | 3.8908 | 5.2293 | 3.4740 | 5.9906 | 2.1698 | 6.2828 | 1.0903 | 5.8454 | 5.4355 | 3.7810 | 4.3097 | 5.6966 | 6.9934 | 2.4912 | 2.5197 | 7.0294 | 5.4764 | 6.8574 | | 1.7669 | 1.7636 | H20 | 2.5888 | 3.8264 | 2.7629 | 4.3733 | 2.1554 | 4.7899 | 1.0890 | 4.6351 | 4.0574 | 3.0996 | 3.7818 | 3.9665 | 5.3704 | 2.5144 | 3.0631 | 5.4764 | 4.0967 | 5.5025 | 1.7669 | | 1.7626 | H21 | 3.3368 | 4.4133 | 2.8053 | 5.3529 | 2.1663 | 6.0531 | 1.0922 | 5.1058 | 4.2873 | 2.6049 | 3.7943 | 5.0369 | 6.2073 | 3.0676 | 2.5079 | 6.8574 | 5.5025 | 6.6344 | 1.7636 | 1.7626 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
108.914 |
|
O1 |
C2 |
H8 |
109.885 |
| O1 |
C2 |
H9 |
109.964 |
|
O1 |
C3 |
C5 |
108.914 |
| O1 |
C3 |
H10 |
109.885 |
|
O1 |
C3 |
H11 |
109.964 |
| C2 |
O1 |
C3 |
112.993 |
|
C2 |
C4 |
C6 |
112.755 |
| C2 |
C4 |
H12 |
108.319 |
|
C2 |
C4 |
H13 |
108.235 |
| C3 |
C5 |
C7 |
112.755 |
|
C3 |
C5 |
H14 |
108.319 |
| C3 |
C5 |
H15 |
108.235 |
|
C4 |
C2 |
H8 |
110.182 |
| C4 |
C2 |
H9 |
110.595 |
|
C4 |
C6 |
H16 |
111.397 |
| C4 |
C6 |
H17 |
110.330 |
|
C4 |
C6 |
H18 |
111.009 |
| C5 |
C3 |
H10 |
110.182 |
|
C5 |
C3 |
H11 |
110.595 |
| C5 |
C7 |
H19 |
111.397 |
|
C5 |
C7 |
H20 |
110.330 |
| C5 |
C7 |
H21 |
111.009 |
|
C6 |
C4 |
H12 |
110.068 |
| C6 |
C4 |
H13 |
110.463 |
|
C7 |
C5 |
H14 |
110.068 |
| C7 |
C5 |
H15 |
110.463 |
|
H8 |
C2 |
H9 |
107.290 |
| H10 |
C3 |
H11 |
107.290 |
|
H12 |
C4 |
H13 |
106.793 |
| H14 |
C5 |
H15 |
106.793 |
|
H16 |
C6 |
H17 |
108.343 |
| H16 |
C6 |
H18 |
107.812 |
|
H17 |
C6 |
H18 |
107.822 |
| H19 |
C7 |
H20 |
108.343 |
|
H19 |
C7 |
H21 |
107.812 |
| H20 |
C7 |
H21 |
107.822 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.361 |
|
|
|
| 2 |
C |
-0.030 |
|
|
|
| 3 |
C |
-0.030 |
|
|
|
| 4 |
C |
-0.293 |
|
|
|
| 5 |
C |
-0.293 |
|
|
|
| 6 |
C |
-0.341 |
|
|
|
| 7 |
C |
-0.341 |
|
|
|
| 8 |
H |
0.102 |
|
|
|
| 9 |
H |
0.110 |
|
|
|
| 10 |
H |
0.102 |
|
|
|
| 11 |
H |
0.110 |
|
|
|
| 12 |
H |
0.136 |
|
|
|
| 13 |
H |
0.123 |
|
|
|
| 14 |
H |
0.136 |
|
|
|
| 15 |
H |
0.123 |
|
|
|
| 16 |
H |
0.120 |
|
|
|
| 17 |
H |
0.139 |
|
|
|
| 18 |
H |
0.113 |
|
|
|
| 19 |
H |
0.120 |
|
|
|
| 20 |
H |
0.139 |
|
|
|
| 21 |
H |
0.113 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.021 |
1.021 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.940 |
0.405 |
0.000 |
| y |
0.405 |
-44.037 |
0.000 |
| z |
0.000 |
0.000 |
-45.876 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.983 |
0.405 |
0.000 |
| y |
0.405 |
2.370 |
0.000 |
| z |
0.000 |
0.000 |
-0.387 |
|
| Polar |
|---|
| 3z2-r2 | -0.774 |
|---|
| x2-y2 | -2.902 |
|---|
| xy | 0.405 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.334 |
-0.514 |
0.000 |
| y |
-0.514 |
12.461 |
0.000 |
| z |
0.000 |
0.000 |
10.491 |
<r2> (average value of r
2) Å
2
| <r2> |
339.647 |
| (<r2>)1/2 |
18.430 |