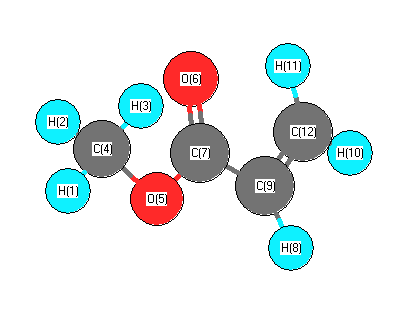
Vibrational Frequencies calculated at B1B95/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3281 |
3138 |
2.04 |
|
|
|
| 2 |
A |
3226 |
3086 |
2.97 |
|
|
|
| 3 |
A |
3199 |
3060 |
13.96 |
|
|
|
| 4 |
A |
3179 |
3041 |
3.72 |
|
|
|
| 5 |
A |
3161 |
3023 |
16.48 |
|
|
|
| 6 |
A |
3077 |
2943 |
30.69 |
|
|
|
| 7 |
A |
1815 |
1736 |
218.59 |
|
|
|
| 8 |
A |
1718 |
1643 |
27.21 |
|
|
|
| 9 |
A |
1495 |
1430 |
7.54 |
|
|
|
| 10 |
A |
1475 |
1411 |
9.76 |
|
|
|
| 11 |
A |
1466 |
1403 |
54.44 |
|
|
|
| 12 |
A |
1431 |
1368 |
70.40 |
|
|
|
| 13 |
A |
1335 |
1277 |
136.68 |
|
|
|
| 14 |
A |
1246 |
1192 |
314.40 |
|
|
|
| 15 |
A |
1198 |
1146 |
10.88 |
|
|
|
| 16 |
A |
1171 |
1120 |
0.78 |
|
|
|
| 17 |
A |
1077 |
1030 |
3.80 |
|
|
|
| 18 |
A |
1053 |
1007 |
14.02 |
|
|
|
| 19 |
A |
1023 |
978 |
36.10 |
|
|
|
| 20 |
A |
1006 |
963 |
21.66 |
|
|
|
| 21 |
A |
879 |
841 |
11.27 |
|
|
|
| 22 |
A |
830 |
794 |
29.10 |
|
|
|
| 23 |
A |
668 |
639 |
5.33 |
|
|
|
| 24 |
A |
530 |
507 |
0.04 |
|
|
|
| 25 |
A |
472 |
452 |
1.52 |
|
|
|
| 26 |
A |
327 |
313 |
16.00 |
|
|
|
| 27 |
A |
209 |
200 |
4.93 |
|
|
|
| 28 |
A |
201 |
192 |
5.06 |
|
|
|
| 29 |
A |
98 |
94 |
3.50 |
|
|
|
| 30 |
A |
49 |
47 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20945.9 cm
-1
Scaled (by 0.9566) Zero Point Vibrational Energy (zpe) 20036.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.160 |
|
|
|
| 2 |
H |
0.174 |
|
|
|
| 3 |
H |
0.174 |
|
|
|
| 4 |
C |
-0.202 |
|
|
|
| 5 |
O |
-0.292 |
|
|
|
| 6 |
O |
-0.488 |
|
|
|
| 7 |
C |
0.186 |
|
|
|
| 8 |
H |
0.164 |
|
|
|
| 9 |
C |
0.141 |
|
|
|
| 10 |
H |
0.158 |
|
|
|
| 11 |
H |
0.183 |
|
|
|
| 12 |
C |
-0.360 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.094 |
-1.568 |
-0.001 |
1.571 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.901 |
-0.736 |
0.000 |
| y |
-0.736 |
-39.102 |
-0.001 |
| z |
0.000 |
-0.001 |
-36.501 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
11.900 |
-0.736 |
0.000 |
| y |
-0.736 |
-7.901 |
-0.001 |
| z |
0.000 |
-0.001 |
-3.999 |
|
| Polar |
|---|
| 3z2-r2 | -7.999 |
|---|
| x2-y2 | 13.201 |
|---|
| xy | -0.736 |
|---|
| xz | 0.000 |
|---|
| yz | -0.001 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.318 |
0.553 |
-0.000 |
| y |
0.553 |
7.653 |
0.000 |
| z |
-0.000 |
0.000 |
5.344 |
<r2> (average value of r
2) Å
2
| <r2> |
176.524 |
| (<r2>)1/2 |
13.286 |