Jump to
S1C2
S1C3
Energy calculated at B1B95/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -581.383505 |
| Energy at 298.15K | |
| HF Energy | -581.383505 |
| Nuclear repulsion energy | 79.198450 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2271 |
2174 |
0.00 |
410.60 |
0.12 |
0.22 |
| 2 |
Ag |
937 |
897 |
0.00 |
10.62 |
0.35 |
0.52 |
| 3 |
Ag |
602 |
577 |
0.00 |
90.65 |
0.21 |
0.35 |
| 4 |
Au |
557 |
533 |
0.00 |
0.00 |
0.00 |
0.00 |
| 5 |
B1u |
2264 |
2167 |
63.59 |
0.00 |
0.00 |
0.00 |
| 6 |
B1u |
855 |
819 |
108.55 |
0.00 |
0.00 |
0.00 |
| 7 |
B2g |
158i |
151i |
0.00 |
23.43 |
0.75 |
0.86 |
| 8 |
B2u |
2301 |
2202 |
99.60 |
0.00 |
0.00 |
0.00 |
| 9 |
B2u |
346 |
331 |
18.07 |
0.00 |
0.00 |
0.00 |
| 10 |
B3g |
2291 |
2193 |
0.00 |
213.77 |
0.75 |
0.86 |
| 11 |
B3g |
591 |
566 |
0.00 |
4.23 |
0.75 |
0.86 |
| 12 |
B3u |
534 |
511 |
1.36 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 6695.7 cm
-1
Scaled (by 0.9571) Zero Point Vibrational Energy (zpe) 6408.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/cc-pVTZ
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
1.063 |
| Si2 |
0.000 |
0.000 |
-1.063 |
| H3 |
0.000 |
1.247 |
1.849 |
| H4 |
0.000 |
-1.247 |
1.849 |
| H5 |
0.000 |
1.247 |
-1.849 |
| H6 |
0.000 |
-1.247 |
-1.849 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1253 | 1.4747 | 1.4747 | 3.1679 | 3.1679 |
Si2 | 2.1253 | | 3.1679 | 3.1679 | 1.4747 | 1.4747 | H3 | 1.4747 | 3.1679 | | 2.4948 | 3.6986 | 4.4614 | H4 | 1.4747 | 3.1679 | 2.4948 | | 4.4614 | 3.6986 | H5 | 3.1679 | 1.4747 | 3.6986 | 4.4614 | | 2.4948 | H6 | 3.1679 | 1.4747 | 4.4614 | 3.6986 | 2.4948 | |
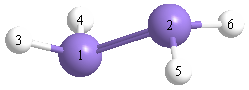 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
122.237 |
|
Si1 |
Si2 |
H6 |
122.237 |
| Si2 |
Si1 |
H3 |
122.237 |
|
Si2 |
Si1 |
H4 |
122.237 |
| H3 |
Si1 |
H4 |
115.526 |
|
H5 |
Si2 |
H6 |
115.526 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
-0.045 |
|
|
|
| 2 |
Si |
-0.045 |
|
|
|
| 3 |
H |
0.022 |
|
|
|
| 4 |
H |
0.022 |
|
|
|
| 5 |
H |
0.022 |
|
|
|
| 6 |
H |
0.022 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.301 |
0.000 |
0.000 |
| y |
0.000 |
-28.991 |
0.000 |
| z |
0.000 |
0.000 |
-27.140 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.236 |
0.000 |
0.000 |
| y |
0.000 |
0.230 |
0.000 |
| z |
0.000 |
0.000 |
3.006 |
|
| Polar |
|---|
| 3z2-r2 | 6.012 |
|---|
| x2-y2 | -2.310 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.895 |
0.000 |
0.000 |
| y |
0.000 |
7.873 |
0.000 |
| z |
0.000 |
0.000 |
14.362 |
<r2> (average value of r
2) Å
2
| <r2> |
69.725 |
| (<r2>)1/2 |
8.350 |
Jump to
S1C1
S1C3
Energy calculated at B1B95/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -581.383845 |
| Energy at 298.15K | |
| HF Energy | -581.383845 |
| Nuclear repulsion energy | 78.815084 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2254 |
2157 |
0.00 |
420.28 |
0.13 |
0.23 |
| 2 |
Ag |
941 |
901 |
0.00 |
16.00 |
0.41 |
0.58 |
| 3 |
Ag |
587 |
561 |
0.00 |
78.14 |
0.23 |
0.38 |
| 4 |
Ag |
233 |
223 |
0.00 |
23.81 |
0.29 |
0.45 |
| 5 |
Au |
2282 |
2184 |
116.13 |
0.00 |
0.00 |
0.00 |
| 6 |
Au |
540 |
517 |
0.00 |
0.00 |
0.00 |
0.00 |
| 7 |
Au |
340 |
325 |
16.86 |
0.00 |
0.00 |
0.00 |
| 8 |
Bg |
2272 |
2174 |
0.00 |
236.22 |
0.75 |
0.86 |
| 9 |
Bg |
599 |
573 |
0.00 |
6.08 |
0.75 |
0.86 |
| 10 |
Bu |
2248 |
2152 |
87.51 |
0.00 |
0.00 |
0.00 |
| 11 |
Bu |
887 |
849 |
146.54 |
0.00 |
0.00 |
0.00 |
| 12 |
Bu |
479 |
458 |
15.54 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 6830.9 cm
-1
Scaled (by 0.9571) Zero Point Vibrational Energy (zpe) 6537.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/cc-pVTZ
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.071 |
0.000 |
| Si2 |
0.000 |
-1.071 |
0.000 |
| H3 |
0.317 |
1.817 |
1.236 |
| H4 |
0.317 |
1.817 |
-1.236 |
| H5 |
-0.317 |
-1.817 |
1.236 |
| H6 |
-0.317 |
-1.817 |
-1.236 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1418 | 1.4780 | 1.4780 | 3.1570 | 3.1570 |
Si2 | 2.1418 | | 3.1570 | 3.1570 | 1.4780 | 1.4780 | H3 | 1.4780 | 3.1570 | | 2.4720 | 3.6883 | 4.4401 | H4 | 1.4780 | 3.1570 | 2.4720 | | 4.4401 | 3.6883 | H5 | 3.1570 | 1.4780 | 3.6883 | 4.4401 | | 2.4720 | H6 | 3.1570 | 1.4780 | 4.4401 | 3.6883 | 2.4720 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
120.302 |
|
Si1 |
Si2 |
H6 |
120.302 |
| Si2 |
Si1 |
H3 |
120.302 |
|
Si2 |
Si1 |
H4 |
120.302 |
| H3 |
Si1 |
H4 |
113.496 |
|
H5 |
Si2 |
H6 |
113.496 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
-0.033 |
|
|
|
| 2 |
Si |
-0.033 |
|
|
|
| 3 |
H |
0.016 |
|
|
|
| 4 |
H |
0.016 |
|
|
|
| 5 |
H |
0.016 |
|
|
|
| 6 |
H |
0.016 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-31.193 |
-0.278 |
0.000 |
| y |
-0.278 |
-27.270 |
0.000 |
| z |
0.000 |
0.000 |
-29.180 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.968 |
-0.278 |
0.000 |
| y |
-0.278 |
2.917 |
0.000 |
| z |
0.000 |
0.000 |
0.051 |
|
| Polar |
|---|
| 3z2-r2 | 0.103 |
|---|
| x2-y2 | -3.923 |
|---|
| xy | -0.278 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.948 |
-0.320 |
0.000 |
| y |
-0.320 |
14.595 |
0.000 |
| z |
0.000 |
0.000 |
8.066 |
<r2> (average value of r
2) Å
2
| <r2> |
70.074 |
| (<r2>)1/2 |
8.371 |
Jump to
S1C1
S1C2
Energy calculated at B1B95/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -581.352405 |
| Energy at 298.15K | |
| HF Energy | -581.352405 |
| Nuclear repulsion energy | 73.560794 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B1B95/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
2111 |
2021 |
311.58 |
264.13 |
0.28 |
0.43 |
| 2 |
A1 |
1637 |
1567 |
0.06 |
67.05 |
0.09 |
0.16 |
| 3 |
A1 |
867 |
830 |
23.09 |
36.43 |
0.31 |
0.47 |
| 4 |
A1 |
390 |
373 |
0.97 |
31.81 |
0.21 |
0.35 |
| 5 |
A1 |
349 |
334 |
0.05 |
16.21 |
0.55 |
0.71 |
| 6 |
A2 |
1434 |
1372 |
0.00 |
0.05 |
0.75 |
0.86 |
| 7 |
A2 |
638 |
610 |
0.00 |
0.27 |
0.75 |
0.86 |
| 8 |
B1 |
1326 |
1269 |
22.19 |
0.20 |
0.75 |
0.86 |
| 9 |
B1 |
829 |
793 |
13.76 |
21.32 |
0.75 |
0.86 |
| 10 |
B2 |
2085 |
1995 |
14.68 |
22.77 |
0.75 |
0.86 |
| 11 |
B2 |
1413 |
1352 |
1034.01 |
12.53 |
0.75 |
0.86 |
| 12 |
B2 |
721 |
690 |
82.71 |
10.44 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 6899.0 cm
-1
Scaled (by 0.9571) Zero Point Vibrational Energy (zpe) 6603.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B1B95/cc-pVTZ
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.306 |
-0.090 |
| Si2 |
0.000 |
-1.306 |
-0.090 |
| H3 |
-1.036 |
0.000 |
-0.157 |
| H4 |
1.036 |
0.000 |
-0.157 |
| H5 |
0.000 |
1.298 |
1.422 |
| H6 |
0.000 |
-1.298 |
1.422 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.6126 | 1.6683 | 1.6683 | 1.5125 | 3.0117 |
Si2 | 2.6126 | | 1.6683 | 1.6683 | 3.0117 | 1.5125 | H3 | 1.6683 | 1.6683 | | 2.0712 | 2.2918 | 2.2918 | H4 | 1.6683 | 1.6683 | 2.0712 | | 2.2918 | 2.2918 | H5 | 1.5125 | 3.0117 | 2.2918 | 2.2918 | | 2.5961 | H6 | 3.0117 | 1.5125 | 2.2918 | 2.2918 | 2.5961 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
30.146 |
|
Si1 |
Si2 |
H6 |
89.688 |
| Si2 |
Si1 |
H3 |
38.464 |
|
Si2 |
Si1 |
H4 |
38.464 |
| H3 |
Si1 |
H4 |
76.738 |
|
H5 |
Si2 |
H6 |
59.542 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B1B95/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
-0.001 |
|
|
|
| 2 |
Si |
-0.001 |
|
|
|
| 3 |
H |
0.005 |
|
|
|
| 4 |
H |
0.005 |
|
|
|
| 5 |
H |
-0.003 |
|
|
|
| 6 |
H |
-0.003 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.421 |
0.421 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-27.095 |
0.000 |
0.000 |
| y |
0.000 |
-31.739 |
0.000 |
| z |
0.000 |
0.000 |
-33.067 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.308 |
0.000 |
0.000 |
| y |
0.000 |
-1.658 |
0.000 |
| z |
0.000 |
0.000 |
-3.650 |
|
| Polar |
|---|
| 3z2-r2 | -7.301 |
|---|
| x2-y2 | 4.644 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.295 |
0.000 |
0.000 |
| y |
0.000 |
13.181 |
0.000 |
| z |
0.000 |
0.000 |
8.812 |
<r2> (average value of r
2) Å
2
| <r2> |
76.751 |
| (<r2>)1/2 |
8.761 |