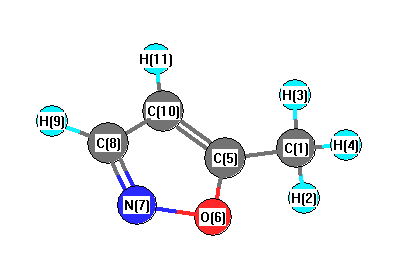
Vibrational Frequencies calculated at B2PLYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3318 |
3149 |
0.11 |
|
|
|
| 2 |
A |
3287 |
3120 |
3.69 |
|
|
|
| 3 |
A |
3187 |
3025 |
8.32 |
|
|
|
| 4 |
A |
3159 |
2998 |
8.06 |
|
|
|
| 5 |
A |
3093 |
2935 |
12.51 |
|
|
|
| 6 |
A |
1660 |
1576 |
31.39 |
|
|
|
| 7 |
A |
1537 |
1459 |
9.56 |
|
|
|
| 8 |
A |
1530 |
1452 |
34.61 |
|
|
|
| 9 |
A |
1524 |
1447 |
7.50 |
|
|
|
| 10 |
A |
1458 |
1384 |
1.62 |
|
|
|
| 11 |
A |
1390 |
1320 |
6.67 |
|
|
|
| 12 |
A |
1283 |
1218 |
19.31 |
|
|
|
| 13 |
A |
1232 |
1170 |
9.40 |
|
|
|
| 14 |
A |
1092 |
1036 |
3.17 |
|
|
|
| 15 |
A |
1074 |
1019 |
3.76 |
|
|
|
| 16 |
A |
1040 |
987 |
5.91 |
|
|
|
| 17 |
A |
1011 |
960 |
3.19 |
|
|
|
| 18 |
A |
940 |
892 |
9.08 |
|
|
|
| 19 |
A |
901 |
856 |
16.05 |
|
|
|
| 20 |
A |
892 |
847 |
6.14 |
|
|
|
| 21 |
A |
796 |
755 |
31.17 |
|
|
|
| 22 |
A |
664 |
630 |
0.55 |
|
|
|
| 23 |
A |
643 |
610 |
3.90 |
|
|
|
| 24 |
A |
638 |
605 |
0.36 |
|
|
|
| 25 |
A |
334 |
317 |
2.02 |
|
|
|
| 26 |
A |
249 |
236 |
1.86 |
|
|
|
| 27 |
A |
99 |
94 |
0.77 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19013.9 cm
-1
Scaled (by 0.9492) Zero Point Vibrational Energy (zpe) 18048.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.