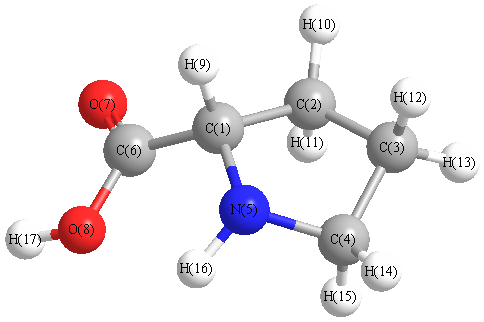
Vibrational Frequencies calculated at B2PLYP/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3756 |
3603 |
72.13 |
|
|
|
| 2 |
A |
3567 |
3423 |
6.60 |
|
|
|
| 3 |
A |
3154 |
3026 |
19.68 |
|
|
|
| 4 |
A |
3127 |
3000 |
13.32 |
|
|
|
| 5 |
A |
3108 |
2982 |
23.58 |
|
|
|
| 6 |
A |
3103 |
2977 |
45.41 |
|
|
|
| 7 |
A |
3084 |
2959 |
19.54 |
|
|
|
| 8 |
A |
3055 |
2931 |
18.11 |
|
|
|
| 9 |
A |
2966 |
2846 |
70.79 |
|
|
|
| 10 |
A |
1802 |
1729 |
232.41 |
|
|
|
| 11 |
A |
1540 |
1478 |
0.55 |
|
|
|
| 12 |
A |
1524 |
1462 |
6.30 |
|
|
|
| 13 |
A |
1505 |
1444 |
1.04 |
|
|
|
| 14 |
A |
1458 |
1398 |
17.71 |
|
|
|
| 15 |
A |
1378 |
1322 |
11.32 |
|
|
|
| 16 |
A |
1367 |
1312 |
9.56 |
|
|
|
| 17 |
A |
1343 |
1288 |
0.99 |
|
|
|
| 18 |
A |
1333 |
1279 |
19.98 |
|
|
|
| 19 |
A |
1328 |
1274 |
3.88 |
|
|
|
| 20 |
A |
1267 |
1216 |
1.39 |
|
|
|
| 21 |
A |
1251 |
1200 |
1.83 |
|
|
|
| 22 |
A |
1222 |
1172 |
11.86 |
|
|
|
| 23 |
A |
1213 |
1164 |
3.13 |
|
|
|
| 24 |
A |
1158 |
1111 |
170.22 |
|
|
|
| 25 |
A |
1147 |
1101 |
83.52 |
|
|
|
| 26 |
A |
1116 |
1070 |
10.60 |
|
|
|
| 27 |
A |
1081 |
1037 |
10.98 |
|
|
|
| 28 |
A |
1001 |
960 |
5.44 |
|
|
|
| 29 |
A |
965 |
926 |
9.47 |
|
|
|
| 30 |
A |
934 |
896 |
2.28 |
|
|
|
| 31 |
A |
918 |
881 |
3.66 |
|
|
|
| 32 |
A |
891 |
855 |
49.02 |
|
|
|
| 33 |
A |
847 |
813 |
28.98 |
|
|
|
| 34 |
A |
786 |
754 |
1.11 |
|
|
|
| 35 |
A |
746 |
716 |
38.77 |
|
|
|
| 36 |
A |
673 |
646 |
85.77 |
|
|
|
| 37 |
A |
624 |
599 |
53.09 |
|
|
|
| 38 |
A |
594 |
570 |
15.21 |
|
|
|
| 39 |
A |
510 |
490 |
33.81 |
|
|
|
| 40 |
A |
495 |
475 |
12.65 |
|
|
|
| 41 |
A |
353 |
339 |
2.10 |
|
|
|
| 42 |
A |
261 |
251 |
2.70 |
|
|
|
| 43 |
A |
185 |
178 |
0.24 |
|
|
|
| 44 |
A |
57 |
55 |
0.17 |
|
|
|
| 45 |
A |
34 |
32 |
1.97 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31912.5 cm
-1
Scaled (by 0.9594) Zero Point Vibrational Energy (zpe) 30616.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.044 |
|
|
|
| 2 |
C |
-0.218 |
|
|
|
| 3 |
C |
-0.202 |
|
|
|
| 4 |
C |
-0.117 |
|
|
|
| 5 |
N |
-0.225 |
|
|
|
| 6 |
C |
0.256 |
|
|
|
| 7 |
O |
-0.290 |
|
|
|
| 8 |
O |
-0.273 |
|
|
|
| 9 |
H |
0.130 |
|
|
|
| 10 |
H |
0.117 |
|
|
|
| 11 |
H |
0.124 |
|
|
|
| 12 |
H |
0.104 |
|
|
|
| 13 |
H |
0.111 |
|
|
|
| 14 |
H |
0.111 |
|
|
|
| 15 |
H |
0.072 |
|
|
|
| 16 |
H |
0.124 |
|
|
|
| 17 |
H |
0.222 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.139 |
-1.306 |
-0.377 |
1.367 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.855 |
5.701 |
-1.139 |
| y |
5.701 |
-50.034 |
2.574 |
| z |
-1.139 |
2.574 |
-47.898 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.111 |
5.701 |
-1.139 |
| y |
5.701 |
-2.657 |
2.574 |
| z |
-1.139 |
2.574 |
0.547 |
|
| Polar |
|---|
| 3z2-r2 | 1.094 |
|---|
| x2-y2 | 3.179 |
|---|
| xy | 5.701 |
|---|
| xz | -1.139 |
|---|
| yz | 2.574 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.033 |
0.049 |
0.043 |
| y |
0.049 |
10.442 |
0.175 |
| z |
0.043 |
0.175 |
8.776 |
<r2> (average value of r
2) Å
2
| <r2> |
258.157 |
| (<r2>)1/2 |
16.067 |