Jump to
S1C2
Energy calculated at B2PLYP/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -358.363366 |
| Energy at 298.15K | -358.368380 |
| HF Energy | -357.964834 |
| Nuclear repulsion energy | 232.565549 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3756 |
3604 |
94.69 |
|
|
|
| 2 |
A' |
3753 |
3600 |
75.11 |
|
|
|
| 3 |
A' |
3611 |
3464 |
62.53 |
|
|
|
| 4 |
A' |
1802 |
1729 |
48.27 |
|
|
|
| 5 |
A' |
1785 |
1713 |
511.93 |
|
|
|
| 6 |
A' |
1605 |
1540 |
108.79 |
|
|
|
| 7 |
A' |
1428 |
1370 |
12.73 |
|
|
|
| 8 |
A' |
1324 |
1270 |
59.13 |
|
|
|
| 9 |
A' |
1184 |
1136 |
284.28 |
|
|
|
| 10 |
A' |
1096 |
1051 |
3.28 |
|
|
|
| 11 |
A' |
779 |
748 |
6.76 |
|
|
|
| 12 |
A' |
612 |
587 |
70.70 |
|
|
|
| 13 |
A' |
528 |
507 |
0.28 |
|
|
|
| 14 |
A' |
415 |
399 |
3.87 |
|
|
|
| 15 |
A' |
271 |
260 |
14.84 |
|
|
|
| 16 |
A" |
837 |
803 |
5.24 |
|
|
|
| 17 |
A" |
674 |
647 |
110.04 |
|
|
|
| 18 |
A" |
632 |
606 |
25.33 |
|
|
|
| 19 |
A" |
435 |
418 |
5.26 |
|
|
|
| 20 |
A" |
306 |
294 |
192.33 |
|
|
|
| 21 |
A" |
63 |
61 |
3.05 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13447.3 cm
-1
Scaled (by 0.9594) Zero Point Vibrational Energy (zpe) 12901.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.752 |
0.000 |
| C2 |
-0.053 |
-0.787 |
0.000 |
| O3 |
-1.093 |
-1.410 |
0.000 |
| O4 |
1.033 |
1.380 |
0.000 |
| O5 |
-1.218 |
1.294 |
0.000 |
| N6 |
1.192 |
-1.308 |
0.000 |
| H7 |
1.306 |
-2.305 |
0.000 |
| H8 |
1.987 |
-0.696 |
0.000 |
| H9 |
-1.090 |
2.254 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5391 | 2.4221 | 1.2097 | 1.3333 | 2.3798 | 3.3240 | 2.4588 | 1.8557 |
C2 | 1.5391 | | 1.2124 | 2.4241 | 2.3840 | 1.3500 | 2.0381 | 2.0426 | 3.2121 | O3 | 2.4221 | 1.2124 | | 3.5082 | 2.7060 | 2.2874 | 2.5613 | 3.1622 | 3.6634 | O4 | 1.2097 | 2.4241 | 3.5082 | | 2.2534 | 2.6932 | 3.6953 | 2.2851 | 2.2957 | O5 | 1.3333 | 2.3840 | 2.7060 | 2.2534 | | 3.5464 | 4.3956 | 3.7728 | 0.9689 | N6 | 2.3798 | 1.3500 | 2.2874 | 2.6932 | 3.5464 | | 1.0031 | 1.0037 | 4.2300 | H7 | 3.3240 | 2.0381 | 2.5613 | 3.6953 | 4.3956 | 1.0031 | | 1.7468 | 5.1499 | H8 | 2.4588 | 2.0426 | 3.1622 | 2.2851 | 3.7728 | 1.0037 | 1.7468 | | 4.2626 | H9 | 1.8557 | 3.2121 | 3.6634 | 2.2957 | 0.9689 | 4.2300 | 5.1499 | 4.2626 | |
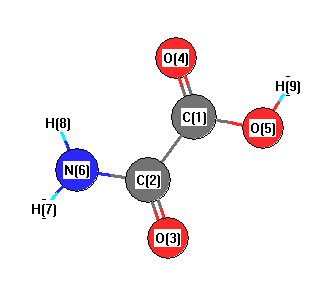 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
122.909 |
|
C1 |
C2 |
N6 |
110.748 |
| C1 |
O5 |
H9 |
106.347 |
|
C2 |
C1 |
O4 |
123.299 |
| C2 |
C1 |
O5 |
111.992 |
|
C2 |
N6 |
H7 |
119.293 |
| C2 |
N6 |
H8 |
119.691 |
|
O3 |
C2 |
N6 |
126.343 |
| O4 |
C1 |
O5 |
124.709 |
|
H7 |
N6 |
H8 |
121.016 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.330 |
|
|
|
| 2 |
C |
0.212 |
|
|
|
| 3 |
O |
-0.327 |
|
|
|
| 4 |
O |
-0.345 |
|
|
|
| 5 |
O |
-0.239 |
|
|
|
| 6 |
N |
-0.247 |
|
|
|
| 7 |
H |
0.184 |
|
|
|
| 8 |
H |
0.198 |
|
|
|
| 9 |
H |
0.235 |
|
|
|
Electric dipole moments
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.247 |
-0.154 |
0.000 |
| y |
-0.154 |
6.628 |
0.000 |
| z |
0.000 |
0.000 |
3.458 |
<r2> (average value of r
2) Å
2
| <r2> |
141.704 |
| (<r2>)1/2 |
11.904 |
Jump to
S1C1
Energy calculated at B2PLYP/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -358.369970 |
| Energy at 298.15K | -358.375264 |
| HF Energy | -357.970802 |
| Nuclear repulsion energy | 234.082206 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3741 |
3589 |
90.84 |
|
|
|
| 2 |
A' |
3600 |
3454 |
92.06 |
|
|
|
| 3 |
A' |
3592 |
3446 |
127.19 |
|
|
|
| 4 |
A' |
1840 |
1765 |
224.78 |
|
|
|
| 5 |
A' |
1774 |
1702 |
303.17 |
|
|
|
| 6 |
A' |
1609 |
1543 |
65.29 |
|
|
|
| 7 |
A' |
1448 |
1389 |
175.56 |
|
|
|
| 8 |
A' |
1351 |
1296 |
361.59 |
|
|
|
| 9 |
A' |
1214 |
1165 |
13.09 |
|
|
|
| 10 |
A' |
1103 |
1058 |
1.96 |
|
|
|
| 11 |
A' |
806 |
773 |
9.83 |
|
|
|
| 12 |
A' |
633 |
607 |
13.07 |
|
|
|
| 13 |
A' |
546 |
523 |
1.96 |
|
|
|
| 14 |
A' |
406 |
389 |
6.84 |
|
|
|
| 15 |
A' |
272 |
261 |
42.38 |
|
|
|
| 16 |
A" |
836 |
803 |
0.03 |
|
|
|
| 17 |
A" |
761 |
730 |
95.13 |
|
|
|
| 18 |
A" |
665 |
638 |
2.62 |
|
|
|
| 19 |
A" |
468 |
449 |
89.42 |
|
|
|
| 20 |
A" |
371 |
356 |
129.08 |
|
|
|
| 21 |
A" |
115 |
111 |
6.22 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13574.6 cm
-1
Scaled (by 0.9594) Zero Point Vibrational Energy (zpe) 13023.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.011 |
-0.793 |
0.000 |
| C2 |
0.000 |
0.746 |
0.000 |
| O3 |
-1.071 |
1.339 |
0.000 |
| O4 |
1.020 |
-1.451 |
0.000 |
| O5 |
-1.222 |
-1.285 |
0.000 |
| N6 |
1.223 |
1.286 |
0.000 |
| H7 |
1.337 |
2.283 |
0.000 |
| H8 |
2.020 |
0.674 |
0.000 |
| H9 |
-1.804 |
-0.499 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5387 | 2.3912 | 1.2042 | 1.3281 | 2.4066 | 3.3499 | 2.4870 | 1.8395 |
C2 | 1.5387 | | 1.2242 | 2.4220 | 2.3700 | 1.3373 | 2.0378 | 2.0212 | 2.1916 | O3 | 2.3912 | 1.2242 | | 3.4867 | 2.6286 | 2.2942 | 2.5861 | 3.1613 | 1.9789 | O4 | 1.2042 | 2.4220 | 3.4867 | | 2.2482 | 2.7451 | 3.7482 | 2.3485 | 2.9806 | O5 | 1.3281 | 2.3700 | 2.6286 | 2.2482 | | 3.5483 | 4.3912 | 3.7878 | 0.9785 | N6 | 2.4066 | 1.3373 | 2.2942 | 2.7451 | 3.5483 | | 1.0036 | 1.0051 | 3.5143 | H7 | 3.3499 | 2.0378 | 2.5861 | 3.7482 | 4.3912 | 1.0036 | | 1.7486 | 4.1961 | H8 | 2.4870 | 2.0212 | 3.1613 | 2.3485 | 3.7878 | 1.0051 | 1.7486 | | 3.9998 | H9 | 1.8395 | 2.1916 | 1.9789 | 2.9806 | 0.9785 | 3.5143 | 4.1961 | 3.9998 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.440 |
|
C1 |
C2 |
N6 |
113.426 |
| C1 |
O5 |
H9 |
104.769 |
|
C2 |
C1 |
O4 |
123.561 |
| C2 |
C1 |
O5 |
111.314 |
|
C2 |
N6 |
H7 |
120.373 |
| C2 |
N6 |
H8 |
118.594 |
|
O3 |
C2 |
N6 |
127.135 |
| O4 |
C1 |
O5 |
125.125 |
|
H7 |
N6 |
H8 |
121.033 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.324 |
|
|
|
| 2 |
C |
0.218 |
|
|
|
| 3 |
O |
-0.381 |
|
|
|
| 4 |
O |
-0.324 |
|
|
|
| 5 |
O |
-0.249 |
|
|
|
| 6 |
N |
-0.228 |
|
|
|
| 7 |
H |
0.186 |
|
|
|
| 8 |
H |
0.204 |
|
|
|
| 9 |
H |
0.250 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.572 |
2.596 |
0.000 |
3.035 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.807 |
7.463 |
0.000 |
| y |
7.463 |
-37.743 |
0.000 |
| z |
0.000 |
0.000 |
-33.410 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.769 |
7.463 |
0.000 |
| y |
7.463 |
-5.634 |
0.000 |
| z |
0.000 |
0.000 |
0.865 |
|
| Polar |
|---|
| 3z2-r2 | 1.731 |
|---|
| x2-y2 | 6.936 |
|---|
| xy | 7.463 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.491 |
0.013 |
0.000 |
| y |
0.013 |
6.236 |
0.000 |
| z |
0.000 |
0.000 |
3.435 |
<r2> (average value of r
2) Å
2
| <r2> |
139.374 |
| (<r2>)1/2 |
11.806 |