Jump to
S1C2
Energy calculated at B2PLYP/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -1755.920315 |
| Energy at 298.15K | -1755.922099 |
| HF Energy | -1755.389647 |
| Nuclear repulsion energy | 700.908961 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
1229 |
1179 |
55.61 |
|
|
|
| 2 |
A |
1198 |
1149 |
196.07 |
|
|
|
| 3 |
A |
1136 |
1089 |
170.47 |
|
|
|
| 4 |
A |
1044 |
1002 |
87.47 |
|
|
|
| 5 |
A |
894 |
858 |
163.39 |
|
|
|
| 6 |
A |
805 |
772 |
279.81 |
|
|
|
| 7 |
A |
658 |
632 |
10.10 |
|
|
|
| 8 |
A |
534 |
513 |
2.77 |
|
|
|
| 9 |
A |
458 |
440 |
1.34 |
|
|
|
| 10 |
A |
438 |
420 |
0.11 |
|
|
|
| 11 |
A |
393 |
377 |
0.13 |
|
|
|
| 12 |
A |
352 |
337 |
0.21 |
|
|
|
| 13 |
A |
315 |
302 |
0.61 |
|
|
|
| 14 |
A |
288 |
276 |
0.07 |
|
|
|
| 15 |
A |
241 |
231 |
0.10 |
|
|
|
| 16 |
A |
200 |
192 |
0.75 |
|
|
|
| 17 |
A |
163 |
157 |
0.51 |
|
|
|
| 18 |
A |
72 |
69 |
0.14 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5208.4 cm
-1
Scaled (by 0.9594) Zero Point Vibrational Energy (zpe) 4996.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/cc-pVTZ
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.593 |
0.223 |
0.270 |
| C2 |
-0.724 |
-0.513 |
-0.200 |
| F3 |
0.484 |
0.294 |
1.631 |
| Cl4 |
1.950 |
-0.877 |
-0.086 |
| Cl5 |
0.788 |
1.710 |
-0.398 |
| Cl6 |
-2.174 |
0.395 |
0.194 |
| F7 |
-0.673 |
-0.711 |
-1.521 |
| F8 |
-0.789 |
-1.710 |
0.391 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
F3 |
Cl4 |
Cl5 |
Cl6 |
F7 |
F8 |
| C1 | | 1.5808 | 1.3669 | 1.7820 | 1.6419 | 2.7738 | 2.3837 | 2.3794 |
C2 | 1.5808 | | 2.3372 | 2.7010 | 2.6962 | 1.7558 | 1.3368 | 1.3360 | F3 | 1.3669 | 2.3372 | | 2.5425 | 2.4930 | 3.0233 | 3.5044 | 2.6781 | Cl4 | 1.7820 | 2.7010 | 2.5425 | | 2.8525 | 4.3246 | 2.9942 | 2.9026 | Cl5 | 1.6419 | 2.6962 | 2.4930 | 2.8525 | | 3.2945 | 3.0419 | 3.8478 | Cl6 | 2.7738 | 1.7558 | 3.0233 | 4.3246 | 3.2945 | | 2.5335 | 2.5273 | F7 | 2.3837 | 1.3368 | 3.5044 | 2.9942 | 3.0419 | 2.5335 | | 2.1606 | F8 | 2.3794 | 1.3360 | 2.6781 | 2.9026 | 3.8478 | 2.5273 | 2.1606 | |
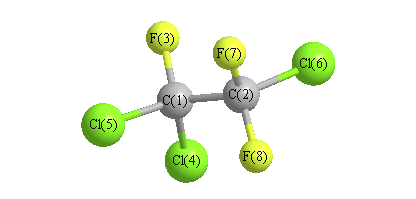 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
Cl6 |
112.367 |
|
C1 |
C2 |
F7 |
109.293 |
| C1 |
C2 |
F8 |
109.037 |
|
C2 |
C1 |
F3 |
104.681 |
| C2 |
C1 |
Cl4 |
106.725 |
|
C2 |
C1 |
Cl5 |
113.558 |
| F3 |
C1 |
Cl4 |
106.953 |
|
F3 |
C1 |
Cl5 |
111.580 |
| Cl4 |
C1 |
Cl5 |
112.777 |
|
Cl6 |
C2 |
F7 |
109.264 |
| Cl6 |
C2 |
F8 |
108.900 |
|
F7 |
C2 |
F8 |
107.875 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.275 |
|
|
|
| 2 |
C |
0.465 |
|
|
|
| 3 |
F |
-0.170 |
|
|
|
| 4 |
Cl |
-0.072 |
|
|
|
| 5 |
Cl |
-0.064 |
|
|
|
| 6 |
Cl |
-0.095 |
|
|
|
| 7 |
F |
-0.169 |
|
|
|
| 8 |
F |
-0.171 |
|
|
|
Electric dipole moments
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.085 |
-1.229 |
-0.630 |
| y |
-1.229 |
9.331 |
-0.215 |
| z |
-0.630 |
-0.215 |
6.978 |
<r2> (average value of r
2) Å
2
| <r2> |
365.924 |
| (<r2>)1/2 |
19.129 |
Jump to
S1C1
Energy calculated at B2PLYP/cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -1755.919610 |
| Energy at 298.15K | -1755.921405 |
| HF Energy | -1755.388703 |
| Nuclear repulsion energy | 702.226037 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B2PLYP/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
1222 |
1172 |
166.38 |
|
|
|
| 2 |
A' |
1108 |
1063 |
7.60 |
|
|
|
| 3 |
A' |
1040 |
998 |
279.57 |
|
|
|
| 4 |
A' |
883 |
847 |
149.49 |
|
|
|
| 5 |
A' |
635 |
609 |
17.29 |
|
|
|
| 6 |
A' |
506 |
486 |
4.23 |
|
|
|
| 7 |
A' |
438 |
420 |
0.08 |
|
|
|
| 8 |
A' |
373 |
358 |
0.19 |
|
|
|
| 9 |
A' |
306 |
293 |
1.48 |
|
|
|
| 10 |
A' |
251 |
241 |
0.03 |
|
|
|
| 11 |
A' |
168 |
161 |
0.25 |
|
|
|
| 12 |
A" |
1181 |
1133 |
138.01 |
|
|
|
| 13 |
A" |
894 |
857 |
236.42 |
|
|
|
| 14 |
A" |
458 |
439 |
2.94 |
|
|
|
| 15 |
A" |
382 |
366 |
0.29 |
|
|
|
| 16 |
A" |
311 |
298 |
0.68 |
|
|
|
| 17 |
A" |
180 |
173 |
0.55 |
|
|
|
| 18 |
A" |
76 |
73 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5204.2 cm
-1
Scaled (by 0.9594) Zero Point Vibrational Energy (zpe) 4992.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B2PLYP/cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.560 |
-0.445 |
0.000 |
| C2 |
-0.174 |
0.924 |
0.000 |
| F3 |
1.904 |
-0.137 |
0.000 |
| Cl4 |
0.251 |
-1.360 |
1.458 |
| Cl5 |
0.251 |
-1.360 |
-1.458 |
| Cl6 |
-1.912 |
0.882 |
0.000 |
| F7 |
0.251 |
1.645 |
1.075 |
| F8 |
0.251 |
1.645 |
-1.075 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
F3 |
Cl4 |
Cl5 |
Cl6 |
F7 |
F8 |
| C1 | | 1.5533 | 1.3792 | 1.7493 | 1.7493 | 2.8059 | 2.3697 | 2.3697 |
C2 | 1.5533 | | 2.3336 | 2.7432 | 2.7432 | 1.7388 | 1.3616 | 1.3616 | F3 | 1.3792 | 2.3336 | | 2.5213 | 2.5213 | 3.9504 | 2.6576 | 2.6576 | Cl4 | 1.7493 | 2.7432 | 2.5213 | | 2.9164 | 3.4400 | 3.0290 | 3.9298 | Cl5 | 1.7493 | 2.7432 | 2.5213 | 2.9164 | | 3.4400 | 3.9298 | 3.0290 | Cl6 | 2.8059 | 1.7388 | 3.9504 | 3.4400 | 3.4400 | | 2.5327 | 2.5327 | F7 | 2.3697 | 1.3616 | 2.6576 | 3.0290 | 3.9298 | 2.5327 | | 2.1493 | F8 | 2.3697 | 1.3616 | 2.6576 | 3.9298 | 3.0290 | 2.5327 | 2.1493 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
Cl6 |
116.811 |
|
C1 |
C2 |
F7 |
108.591 |
| C1 |
C2 |
F8 |
108.591 |
|
C2 |
C1 |
F3 |
105.300 |
| C2 |
C1 |
Cl4 |
112.192 |
|
C2 |
C1 |
Cl5 |
112.192 |
| F3 |
C1 |
Cl4 |
106.803 |
|
F3 |
C1 |
Cl5 |
106.803 |
| Cl4 |
C1 |
Cl5 |
112.941 |
|
Cl6 |
C2 |
F7 |
108.942 |
| Cl6 |
C2 |
F8 |
108.942 |
|
F7 |
C2 |
F8 |
104.231 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B2PLYP/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.273 |
|
|
|
| 2 |
C |
0.459 |
|
|
|
| 3 |
F |
-0.172 |
|
|
|
| 4 |
Cl |
-0.065 |
|
|
|
| 5 |
Cl |
-0.065 |
|
|
|
| 6 |
Cl |
-0.089 |
|
|
|
| 7 |
F |
-0.171 |
|
|
|
| 8 |
F |
-0.171 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.651 |
-0.621 |
0.000 |
0.900 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-63.681 |
-0.161 |
0.000 |
| y |
-0.161 |
-64.055 |
0.000 |
| z |
0.000 |
0.000 |
-63.027 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.140 |
-0.161 |
0.000 |
| y |
-0.161 |
-0.701 |
0.000 |
| z |
0.000 |
0.000 |
0.841 |
|
| Polar |
|---|
| 3z2-r2 | 1.682 |
|---|
| x2-y2 | 0.374 |
|---|
| xy | -0.161 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.546 |
-0.054 |
0.000 |
| y |
-0.054 |
8.059 |
0.000 |
| z |
0.000 |
0.000 |
9.337 |
<r2> (average value of r
2) Å
2
| <r2> |
359.266 |
| (<r2>)1/2 |
18.954 |