Jump to
S1C2
Energy calculated at HSEh1PBE/6-311G**
| | hartrees |
|---|
| Energy at 0K | -750.916287 |
| Energy at 298.15K | |
| HF Energy | -750.916287 |
| Nuclear repulsion energy | 564.181911 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HSEh1PBE/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
1864 |
1790 |
0.00 |
|
|
|
| 2 |
Ag |
1453 |
1396 |
0.00 |
|
|
|
| 3 |
Ag |
1351 |
1298 |
0.00 |
|
|
|
| 4 |
Ag |
1206 |
1158 |
0.00 |
|
|
|
| 5 |
Ag |
730 |
701 |
0.00 |
|
|
|
| 6 |
Ag |
596 |
572 |
0.00 |
|
|
|
| 7 |
Ag |
383 |
368 |
0.00 |
|
|
|
| 8 |
Ag |
355 |
341 |
0.00 |
|
|
|
| 9 |
Ag |
221 |
212 |
0.00 |
|
|
|
| 10 |
Au |
576 |
553 |
2.34 |
|
|
|
| 11 |
Au |
350 |
336 |
3.70 |
|
|
|
| 12 |
Au |
111 |
107 |
0.03 |
|
|
|
| 13 |
Au |
11i |
10i |
0.11 |
|
|
|
| 14 |
Bg |
652 |
626 |
0.00 |
|
|
|
| 15 |
Bg |
481 |
462 |
0.00 |
|
|
|
| 16 |
Bg |
181 |
174 |
0.00 |
|
|
|
| 17 |
Bu |
1806 |
1735 |
428.06 |
|
|
|
| 18 |
Bu |
1370 |
1316 |
465.51 |
|
|
|
| 19 |
Bu |
1236 |
1187 |
303.05 |
|
|
|
| 20 |
Bu |
976 |
938 |
308.76 |
|
|
|
| 21 |
Bu |
630 |
605 |
3.78 |
|
|
|
| 22 |
Bu |
501 |
482 |
3.40 |
|
|
|
| 23 |
Bu |
290 |
278 |
5.03 |
|
|
|
| 24 |
Bu |
137 |
132 |
0.77 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8722.5 cm
-1
Scaled (by 0.9604) Zero Point Vibrational Energy (zpe) 8377.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HSEh1PBE/6-311G**
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.153 |
1.850 |
0.000 |
| C2 |
0.465 |
0.553 |
0.000 |
| C3 |
-0.465 |
-0.553 |
0.000 |
| C4 |
-0.153 |
-1.850 |
0.000 |
| F5 |
1.062 |
2.792 |
0.000 |
| F6 |
-1.062 |
2.333 |
0.000 |
| F7 |
1.764 |
0.229 |
0.000 |
| F8 |
-1.764 |
-0.229 |
0.000 |
| F9 |
1.062 |
-2.333 |
0.000 |
| F10 |
-1.062 |
-2.792 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| C1 | | 1.3336 | 2.4813 | 3.7121 | 1.3097 | 1.3077 | 2.2857 | 2.8279 | 4.2808 | 4.7986 |
C2 | 1.3336 | | 1.4457 | 2.4813 | 2.3174 | 2.3455 | 1.3391 | 2.3629 | 2.9477 | 3.6779 | C3 | 2.4813 | 1.4457 | | 1.3336 | 3.6779 | 2.9477 | 2.3629 | 1.3391 | 2.3455 | 2.3174 | C4 | 3.7121 | 2.4813 | 1.3336 | | 4.7986 | 4.2808 | 2.8279 | 2.2857 | 1.3077 | 1.3097 | F5 | 1.3097 | 2.3174 | 3.6779 | 4.7986 | | 2.1733 | 2.6581 | 4.1374 | 5.1258 | 5.9753 | F6 | 1.3077 | 2.3455 | 2.9477 | 4.2808 | 2.1733 | | 3.5240 | 2.6567 | 5.1274 | 5.1258 | F7 | 2.2857 | 1.3391 | 2.3629 | 2.8279 | 2.6581 | 3.5240 | | 3.5585 | 2.6567 | 4.1374 | F8 | 2.8279 | 2.3629 | 1.3391 | 2.2857 | 4.1374 | 2.6567 | 3.5585 | | 3.5240 | 2.6581 | F9 | 4.2808 | 2.9477 | 2.3455 | 1.3077 | 5.1258 | 5.1274 | 2.6567 | 3.5240 | | 2.1733 | F10 | 4.7986 | 3.6779 | 2.3174 | 1.3097 | 5.9753 | 5.1258 | 4.1374 | 2.6581 | 2.1733 | |
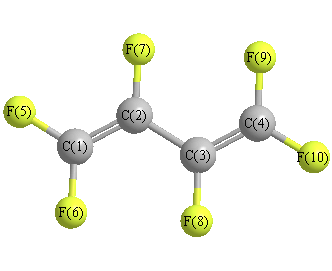 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
126.399 |
|
C1 |
C2 |
F7 |
117.564 |
| C2 |
C1 |
F5 |
122.493 |
|
C2 |
C1 |
F6 |
125.247 |
| C2 |
C3 |
C4 |
126.399 |
|
C2 |
C3 |
F8 |
116.037 |
| C3 |
C2 |
F7 |
116.037 |
|
C3 |
C4 |
F9 |
125.247 |
| C3 |
C4 |
F10 |
122.493 |
|
C4 |
C3 |
F8 |
117.564 |
| F5 |
C1 |
F6 |
112.260 |
|
F9 |
C4 |
F10 |
112.260 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.469 |
|
|
|
| 2 |
C |
0.040 |
|
|
|
| 3 |
C |
0.040 |
|
|
|
| 4 |
C |
0.469 |
|
|
|
| 5 |
F |
-0.157 |
|
|
|
| 6 |
F |
-0.151 |
|
|
|
| 7 |
F |
-0.201 |
|
|
|
| 8 |
F |
-0.201 |
|
|
|
| 9 |
F |
-0.151 |
|
|
|
| 10 |
F |
-0.157 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-55.169 |
-0.267 |
0.000 |
| y |
-0.267 |
-52.250 |
0.000 |
| z |
0.000 |
0.000 |
-49.194 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.447 |
-0.267 |
0.000 |
| y |
-0.267 |
-0.068 |
0.000 |
| z |
0.000 |
0.000 |
4.515 |
|
| Polar |
|---|
| 3z2-r2 | 9.030 |
|---|
| x2-y2 | -2.919 |
|---|
| xy | -0.267 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.036 |
-0.082 |
0.000 |
| y |
-0.082 |
11.862 |
0.000 |
| z |
0.000 |
0.000 |
3.423 |
<r2> (average value of r
2) Å
2
| <r2> |
416.175 |
| (<r2>)1/2 |
20.400 |
Jump to
S1C1
Energy calculated at HSEh1PBE/6-311G**
| | hartrees |
|---|
| Energy at 0K | -750.917393 |
| Energy at 298.15K | -750.918628 |
| HF Energy | -750.917393 |
| Nuclear repulsion energy | 568.229951 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at HSEh1PBE/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
1860 |
1786 |
74.10 |
|
|
|
| 2 |
A |
1419 |
1362 |
13.68 |
|
|
|
| 3 |
A |
1365 |
1311 |
192.49 |
|
|
|
| 4 |
A |
1163 |
1117 |
312.20 |
|
|
|
| 5 |
A |
729 |
701 |
0.24 |
|
|
|
| 6 |
A |
676 |
650 |
1.22 |
|
|
|
| 7 |
A |
543 |
522 |
0.02 |
|
|
|
| 8 |
A |
479 |
460 |
0.14 |
|
|
|
| 9 |
A |
384 |
369 |
0.71 |
|
|
|
| 10 |
A |
259 |
248 |
0.40 |
|
|
|
| 11 |
A |
186 |
179 |
0.41 |
|
|
|
| 12 |
A |
100 |
96 |
0.22 |
|
|
|
| 13 |
A |
51 |
49 |
0.00 |
|
|
|
| 14 |
B |
1833 |
1760 |
301.10 |
|
|
|
| 15 |
B |
1362 |
1308 |
302.81 |
|
|
|
| 16 |
B |
1220 |
1171 |
114.96 |
|
|
|
| 17 |
B |
997 |
958 |
206.31 |
|
|
|
| 18 |
B |
637 |
611 |
2.87 |
|
|
|
| 19 |
B |
630 |
605 |
5.69 |
|
|
|
| 20 |
B |
570 |
547 |
3.48 |
|
|
|
| 21 |
B |
422 |
405 |
3.64 |
|
|
|
| 22 |
B |
298 |
287 |
3.97 |
|
|
|
| 23 |
B |
209 |
201 |
3.12 |
|
|
|
| 24 |
B |
109 |
104 |
0.34 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8749.2 cm
-1
Scaled (by 0.9604) Zero Point Vibrational Energy (zpe) 8402.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at HSEh1PBE/6-311G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.087 |
1.558 |
-0.374 |
| C2 |
0.264 |
0.672 |
0.601 |
| C3 |
-0.264 |
-0.672 |
0.601 |
| C4 |
-0.087 |
-1.558 |
-0.374 |
| F5 |
0.617 |
2.752 |
-0.380 |
| F6 |
-0.617 |
1.325 |
-1.454 |
| F7 |
0.969 |
1.041 |
1.683 |
| F8 |
-0.969 |
-1.041 |
1.683 |
| F9 |
0.617 |
-1.325 |
-1.454 |
| F10 |
-0.617 |
-2.752 |
-0.380 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
| C1 | | 1.3292 | 2.4592 | 3.1216 | 1.3058 | 1.3099 | 2.2967 | 3.4791 | 3.1241 | 4.3675 |
C2 | 1.3292 | | 1.4438 | 2.4592 | 2.3264 | 2.3285 | 1.3432 | 2.3718 | 2.8867 | 3.6688 | C3 | 2.4592 | 1.4438 | | 1.3292 | 3.6688 | 2.8867 | 2.3718 | 1.3432 | 2.3285 | 2.3264 | C4 | 3.1216 | 2.4592 | 1.3292 | | 4.3675 | 3.1241 | 3.4791 | 2.2967 | 1.3099 | 1.3058 | F5 | 1.3058 | 2.3264 | 3.6688 | 4.3675 | | 2.1707 | 2.7026 | 4.5997 | 4.2161 | 5.6406 | F6 | 1.3099 | 2.3285 | 2.8867 | 3.1241 | 2.1707 | | 3.5258 | 3.9446 | 2.9234 | 4.2161 | F7 | 2.2967 | 1.3432 | 2.3718 | 3.4791 | 2.7026 | 3.5258 | | 2.8445 | 3.9446 | 4.5997 | F8 | 3.4791 | 2.3718 | 1.3432 | 2.2967 | 4.5997 | 3.9446 | 2.8445 | | 3.5258 | 2.7026 | F9 | 3.1241 | 2.8867 | 2.3285 | 1.3099 | 4.2161 | 2.9234 | 3.9446 | 3.5258 | | 2.1707 | F10 | 4.3675 | 3.6688 | 2.3264 | 1.3058 | 5.6406 | 4.2161 | 4.5997 | 2.7026 | 2.1707 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
124.905 |
|
C1 |
C2 |
F7 |
118.492 |
| C2 |
C1 |
F5 |
123.980 |
|
C2 |
C1 |
F6 |
123.843 |
| C2 |
C3 |
C4 |
124.905 |
|
C2 |
C3 |
F8 |
116.597 |
| C3 |
C2 |
F7 |
116.597 |
|
C3 |
C4 |
F9 |
123.843 |
| C3 |
C4 |
F10 |
123.980 |
|
C4 |
C3 |
F8 |
118.492 |
| F5 |
C1 |
F6 |
112.165 |
|
F9 |
C4 |
F10 |
112.165 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at HSEh1PBE/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.425 |
|
|
|
| 2 |
C |
0.081 |
|
|
|
| 3 |
C |
0.081 |
|
|
|
| 4 |
C |
0.425 |
|
|
|
| 5 |
F |
-0.149 |
|
|
|
| 6 |
F |
-0.156 |
|
|
|
| 7 |
F |
-0.201 |
|
|
|
| 8 |
F |
-0.201 |
|
|
|
| 9 |
F |
-0.156 |
|
|
|
| 10 |
F |
-0.149 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.950 |
0.950 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-51.049 |
-1.658 |
0.000 |
| y |
-1.658 |
-51.983 |
0.000 |
| z |
0.000 |
0.000 |
-54.073 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.979 |
-1.658 |
0.000 |
| y |
-1.658 |
0.578 |
0.000 |
| z |
0.000 |
0.000 |
-2.557 |
|
| Polar |
|---|
| 3z2-r2 | -5.113 |
|---|
| x2-y2 | 0.934 |
|---|
| xy | -1.658 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.311 |
0.685 |
0.000 |
| y |
0.685 |
8.842 |
0.000 |
| z |
0.000 |
0.000 |
6.719 |
<r2> (average value of r
2) Å
2
| <r2> |
383.841 |
| (<r2>)1/2 |
19.592 |