Vibrational Frequencies calculated at HSEh1PBE/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3162 |
3019 |
7.48 |
|
|
|
| 2 |
A |
3153 |
3010 |
5.54 |
|
|
|
| 3 |
A |
3135 |
2993 |
16.31 |
|
|
|
| 4 |
A |
3091 |
2951 |
18.21 |
|
|
|
| 5 |
A |
3081 |
2942 |
23.65 |
|
|
|
| 6 |
A |
3064 |
2926 |
5.83 |
|
|
|
| 7 |
A |
1832 |
1749 |
433.18 |
|
|
|
| 8 |
A |
1502 |
1434 |
1.01 |
|
|
|
| 9 |
A |
1486 |
1419 |
8.70 |
|
|
|
| 10 |
A |
1457 |
1391 |
10.03 |
|
|
|
| 11 |
A |
1364 |
1302 |
0.08 |
|
|
|
| 12 |
A |
1319 |
1259 |
6.11 |
|
|
|
| 13 |
A |
1313 |
1253 |
5.85 |
|
|
|
| 14 |
A |
1252 |
1195 |
2.11 |
|
|
|
| 15 |
A |
1198 |
1144 |
1.38 |
|
|
|
| 16 |
A |
1122 |
1071 |
13.89 |
|
|
|
| 17 |
A |
1087 |
1038 |
63.43 |
|
|
|
| 18 |
A |
1044 |
996 |
42.05 |
|
|
|
| 19 |
A |
1037 |
990 |
1.09 |
|
|
|
| 20 |
A |
935 |
893 |
8.43 |
|
|
|
| 21 |
A |
878 |
838 |
8.58 |
|
|
|
| 22 |
A |
845 |
807 |
11.81 |
|
|
|
| 23 |
A |
713 |
681 |
1.95 |
|
|
|
| 24 |
A |
642 |
613 |
39.25 |
|
|
|
| 25 |
A |
606 |
579 |
10.21 |
|
|
|
| 26 |
A |
486 |
464 |
0.64 |
|
|
|
| 27 |
A |
481 |
459 |
2.14 |
|
|
|
| 28 |
A |
408 |
390 |
3.52 |
|
|
|
| 29 |
A |
231 |
220 |
1.35 |
|
|
|
| 30 |
A |
133 |
127 |
0.85 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21026.5 cm
-1
Scaled (by 0.9548) Zero Point Vibrational Energy (zpe) 20076.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
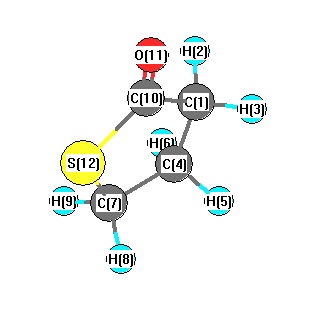
Charges from optimized geometry at HSEh1PBE/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.295 |
|
|
|
| 2 |
H |
0.205 |
|
|
|
| 3 |
H |
0.208 |
|
|
|
| 4 |
C |
-0.324 |
|
|
|
| 5 |
H |
0.181 |
|
|
|
| 6 |
H |
0.194 |
|
|
|
| 7 |
C |
-0.476 |
|
|
|
| 8 |
H |
0.211 |
|
|
|
| 9 |
H |
0.207 |
|
|
|
| 10 |
C |
0.073 |
|
|
|
| 11 |
O |
-0.382 |
|
|
|
| 12 |
S |
0.198 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
4.082 |
1.764 |
0.396 |
4.464 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.982 |
-0.216 |
-0.038 |
| y |
-0.216 |
-42.581 |
0.138 |
| z |
-0.038 |
0.138 |
-42.457 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-5.463 |
-0.216 |
-0.038 |
| y |
-0.216 |
2.639 |
0.138 |
| z |
-0.038 |
0.138 |
2.824 |
|
| Polar |
|---|
| 3z2-r2 | 5.649 |
|---|
| x2-y2 | -5.402 |
|---|
| xy | -0.216 |
|---|
| xz | -0.038 |
|---|
| yz | 0.138 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.206 |
-0.494 |
0.110 |
| y |
-0.494 |
10.435 |
0.127 |
| z |
0.110 |
0.127 |
7.404 |
<r2> (average value of r
2) Å
2
| <r2> |
178.297 |
| (<r2>)1/2 |
13.353 |