Vibrational Frequencies calculated at HSEh1PBE/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3697 |
3548 |
75.81 |
|
|
|
| 2 |
A' |
3262 |
3130 |
1.83 |
|
|
|
| 3 |
A' |
3203 |
3074 |
20.03 |
|
|
|
| 4 |
A' |
3189 |
3060 |
18.95 |
|
|
|
| 5 |
A' |
1688 |
1620 |
68.38 |
|
|
|
| 6 |
A' |
1648 |
1582 |
85.40 |
|
|
|
| 7 |
A' |
1554 |
1491 |
28.57 |
|
|
|
| 8 |
A' |
1509 |
1448 |
2.55 |
|
|
|
| 9 |
A' |
1454 |
1395 |
52.53 |
|
|
|
| 10 |
A' |
1442 |
1384 |
37.07 |
|
|
|
| 11 |
A' |
1387 |
1331 |
65.28 |
|
|
|
| 12 |
A' |
1331 |
1278 |
12.91 |
|
|
|
| 13 |
A' |
1324 |
1271 |
16.73 |
|
|
|
| 14 |
A' |
1297 |
1245 |
33.11 |
|
|
|
| 15 |
A' |
1219 |
1170 |
6.97 |
|
|
|
| 16 |
A' |
1152 |
1105 |
7.08 |
|
|
|
| 17 |
A' |
1101 |
1057 |
16.19 |
|
|
|
| 18 |
A' |
956 |
917 |
1.51 |
|
|
|
| 19 |
A' |
922 |
885 |
12.40 |
|
|
|
| 20 |
A' |
818 |
785 |
14.18 |
|
|
|
| 21 |
A' |
666 |
639 |
0.34 |
|
|
|
| 22 |
A' |
574 |
551 |
3.82 |
|
|
|
| 23 |
A' |
448 |
430 |
13.10 |
|
|
|
| 24 |
A" |
999 |
959 |
0.34 |
|
|
|
| 25 |
A" |
947 |
909 |
11.44 |
|
|
|
| 26 |
A" |
883 |
847 |
5.47 |
|
|
|
| 27 |
A" |
826 |
793 |
12.49 |
|
|
|
| 28 |
A" |
674 |
647 |
4.83 |
|
|
|
| 29 |
A" |
631 |
606 |
31.10 |
|
|
|
| 30 |
A" |
560 |
537 |
105.23 |
|
|
|
| 31 |
A" |
423 |
405 |
3.57 |
|
|
|
| 32 |
A" |
249 |
239 |
0.42 |
|
|
|
| 33 |
A" |
233 |
224 |
5.39 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21132.1 cm
-1
Scaled (by 0.9596) Zero Point Vibrational Energy (zpe) 20278.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
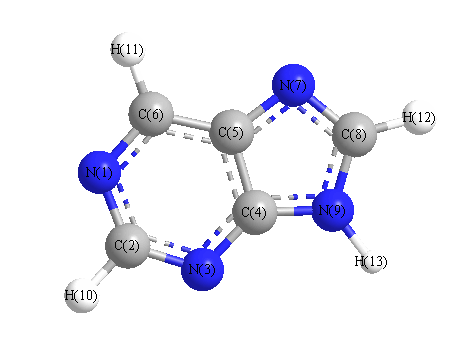
Charges from optimized geometry at HSEh1PBE/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.319 |
|
|
|
| 2 |
C |
0.012 |
|
|
|
| 3 |
N |
-0.335 |
|
|
|
| 4 |
C |
0.471 |
|
|
|
| 5 |
C |
-0.063 |
|
|
|
| 6 |
C |
0.020 |
|
|
|
| 7 |
N |
-0.341 |
|
|
|
| 8 |
C |
0.098 |
|
|
|
| 9 |
N |
-0.612 |
|
|
|
| 10 |
H |
0.221 |
|
|
|
| 11 |
H |
0.236 |
|
|
|
| 12 |
H |
0.238 |
|
|
|
| 13 |
H |
0.374 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.234 |
3.025 |
0.000 |
3.761 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-49.919 |
6.258 |
0.000 |
| y |
6.258 |
-46.834 |
0.000 |
| z |
0.000 |
0.000 |
-51.851 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.576 |
6.258 |
0.000 |
| y |
6.258 |
4.051 |
0.000 |
| z |
0.000 |
0.000 |
-3.474 |
|
| Polar |
|---|
| 3z2-r2 | -6.949 |
|---|
| x2-y2 | -3.085 |
|---|
| xy | 6.258 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
14.917 |
1.372 |
0.000 |
| y |
1.372 |
11.627 |
0.000 |
| z |
0.000 |
0.000 |
5.026 |
<r2> (average value of r
2) Å
2
| <r2> |
252.199 |
| (<r2>)1/2 |
15.881 |