Jump to
S1C2
Energy calculated at M06-2X/6-311+G(3df,2p)
| | hartrees |
|---|
| Energy at 0K | -303.076518 |
| Energy at 298.15K | |
| HF Energy | -303.076518 |
| Nuclear repulsion energy | 160.562508 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/6-311+G(3df,2p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3055 |
3055 |
59.35 |
109.16 |
0.24 |
0.39 |
| 2 |
A1 |
1967 |
1967 |
35.39 |
45.04 |
0.15 |
0.26 |
| 3 |
A1 |
1470 |
1470 |
0.23 |
3.05 |
0.45 |
0.62 |
| 4 |
A1 |
1182 |
1182 |
111.32 |
2.59 |
0.16 |
0.28 |
| 5 |
A1 |
557 |
557 |
0.96 |
7.33 |
0.22 |
0.36 |
| 6 |
A1 |
289 |
289 |
14.56 |
0.65 |
0.38 |
0.55 |
| 7 |
A2 |
1042 |
1042 |
0.00 |
0.75 |
0.75 |
0.86 |
| 8 |
A2 |
184 |
184 |
0.00 |
0.47 |
0.75 |
0.86 |
| 9 |
B1 |
1051 |
1051 |
0.19 |
1.36 |
0.75 |
0.86 |
| 10 |
B1 |
130 |
130 |
4.80 |
0.08 |
0.75 |
0.86 |
| 11 |
B2 |
3032 |
3032 |
2.14 |
0.70 |
0.75 |
0.86 |
| 12 |
B2 |
1886 |
1886 |
762.47 |
2.74 |
0.75 |
0.86 |
| 13 |
B2 |
1408 |
1408 |
11.30 |
4.45 |
0.75 |
0.86 |
| 14 |
B2 |
1139 |
1139 |
695.69 |
1.74 |
0.75 |
0.86 |
| 15 |
B2 |
724 |
724 |
50.89 |
0.05 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 9558.0 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 9558.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/6-311+G(3df,2p)
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.379 |
| C2 |
0.000 |
1.172 |
-0.320 |
| C3 |
0.000 |
-1.172 |
-0.320 |
| O4 |
0.000 |
2.218 |
0.226 |
| O5 |
0.000 |
-2.218 |
0.226 |
| H6 |
0.000 |
1.034 |
-1.410 |
| H7 |
0.000 |
-1.034 |
-1.410 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
O4 |
O5 |
H6 |
H7 |
| O1 | | 1.3645 | 1.3645 | 2.2228 | 2.2228 | 2.0665 | 2.0665 |
C2 | 1.3645 | | 2.3442 | 1.1794 | 3.4334 | 1.0993 | 2.4611 | C3 | 1.3645 | 2.3442 | | 3.4334 | 1.1794 | 2.4611 | 1.0993 | O4 | 2.2228 | 1.1794 | 3.4334 | | 4.4351 | 2.0196 | 3.6404 | O5 | 2.2228 | 3.4334 | 1.1794 | 4.4351 | | 3.6404 | 2.0196 | H6 | 2.0665 | 1.0993 | 2.4611 | 2.0196 | 3.6404 | | 2.0684 | H7 | 2.0665 | 2.4611 | 1.0993 | 3.6404 | 2.0196 | 2.0684 | |
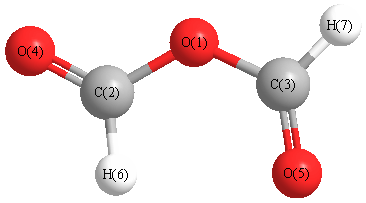 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
O4 |
121.631 |
|
O1 |
C2 |
H6 |
113.585 |
| O1 |
C3 |
O5 |
121.631 |
|
C2 |
O1 |
C3 |
118.413 |
| O4 |
C2 |
H6 |
124.784 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-311+G(3df,2p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.562 |
|
|
|
| 2 |
C |
0.764 |
|
|
|
| 3 |
C |
0.764 |
|
|
|
| 4 |
O |
-0.661 |
|
|
|
| 5 |
O |
-0.661 |
|
|
|
| 6 |
H |
0.178 |
|
|
|
| 7 |
H |
0.178 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-3.483 |
3.483 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-26.541 |
0.000 |
0.000 |
| y |
0.000 |
-39.331 |
0.000 |
| z |
0.000 |
0.000 |
-25.984 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.117 |
0.000 |
0.000 |
| y |
0.000 |
-13.069 |
0.000 |
| z |
0.000 |
0.000 |
6.952 |
|
| Polar |
|---|
| 3z2-r2 | 13.903 |
|---|
| x2-y2 | 12.791 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.282 |
0.000 |
0.000 |
| y |
0.000 |
7.546 |
0.000 |
| z |
0.000 |
0.000 |
4.663 |
<r2> (average value of r
2) Å
2
| <r2> |
123.603 |
| (<r2>)1/2 |
11.118 |
Jump to
S1C1
Energy calculated at M06-2X/6-311+G(3df,2p)
| | hartrees |
|---|
| Energy at 0K | -303.081644 |
| Energy at 298.15K | |
| HF Energy | -303.081644 |
| Nuclear repulsion energy | 163.106112 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/6-311+G(3df,2p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3127 |
3127 |
6.52 |
33.55 |
0.28 |
0.44 |
| 2 |
A' |
3105 |
3105 |
24.70 |
115.05 |
0.24 |
0.39 |
| 3 |
A' |
1927 |
1927 |
165.07 |
32.70 |
0.14 |
0.24 |
| 4 |
A' |
1875 |
1875 |
481.05 |
4.87 |
0.34 |
0.51 |
| 5 |
A' |
1425 |
1425 |
2.73 |
3.04 |
0.31 |
0.47 |
| 6 |
A' |
1409 |
1409 |
1.61 |
5.35 |
0.74 |
0.85 |
| 7 |
A' |
1175 |
1175 |
670.22 |
2.49 |
0.16 |
0.28 |
| 8 |
A' |
1059 |
1059 |
210.42 |
3.72 |
0.08 |
0.14 |
| 9 |
A' |
808 |
808 |
0.61 |
3.66 |
0.44 |
0.61 |
| 10 |
A' |
559 |
559 |
6.05 |
2.82 |
0.27 |
0.42 |
| 11 |
A' |
272 |
272 |
11.43 |
1.41 |
0.33 |
0.49 |
| 12 |
A" |
1071 |
1071 |
0.00 |
2.00 |
0.75 |
0.86 |
| 13 |
A" |
1061 |
1061 |
0.10 |
0.21 |
0.75 |
0.86 |
| 14 |
A" |
224 |
224 |
12.79 |
0.86 |
0.75 |
0.86 |
| 15 |
A" |
81 |
81 |
40.27 |
0.85 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 9589.2 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 9589.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/6-311+G(3df,2p)
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.711 |
0.000 |
| C2 |
0.975 |
-0.263 |
0.000 |
| C3 |
-1.306 |
0.327 |
0.000 |
| O4 |
2.115 |
0.041 |
0.000 |
| O5 |
-1.696 |
-0.793 |
0.000 |
| H6 |
0.571 |
-1.279 |
0.000 |
| H7 |
-1.937 |
1.222 |
0.000 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
O4 |
O5 |
H6 |
H7 |
| O1 | | 1.3782 | 1.3611 | 2.2187 | 2.2666 | 2.0702 | 2.0028 |
C2 | 1.3782 | | 2.3559 | 1.1801 | 2.7228 | 1.0929 | 3.2682 | C3 | 1.3611 | 2.3559 | | 3.4329 | 1.1859 | 2.4702 | 1.0945 | O4 | 2.2187 | 1.1801 | 3.4329 | | 3.9012 | 2.0313 | 4.2202 | O5 | 2.2666 | 2.7228 | 1.1859 | 3.9012 | | 2.3186 | 2.0287 | H6 | 2.0702 | 1.0929 | 2.4702 | 2.0313 | 2.3186 | | 3.5413 | H7 | 2.0028 | 3.2682 | 1.0945 | 4.2202 | 2.0287 | 3.5413 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
O4 |
120.075 |
|
O1 |
C2 |
H6 |
113.301 |
| O1 |
C3 |
O5 |
125.587 |
|
C2 |
O1 |
C3 |
118.637 |
| O4 |
C2 |
H6 |
126.624 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-311+G(3df,2p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.544 |
|
|
|
| 2 |
C |
0.722 |
|
|
|
| 3 |
C |
0.727 |
|
|
|
| 4 |
O |
-0.675 |
|
|
|
| 5 |
O |
-0.649 |
|
|
|
| 6 |
H |
0.230 |
|
|
|
| 7 |
H |
0.188 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.853 |
0.522 |
0.000 |
1.926 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-34.309 |
-5.710 |
0.000 |
| y |
-5.710 |
-27.764 |
0.000 |
| z |
0.000 |
0.000 |
-26.551 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-7.152 |
-5.710 |
0.000 |
| y |
-5.710 |
2.666 |
0.000 |
| z |
0.000 |
0.000 |
4.486 |
|
| Polar |
|---|
| 3z2-r2 | 8.972 |
|---|
| x2-y2 | -6.546 |
|---|
| xy | -5.710 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.854 |
0.208 |
0.000 |
| y |
0.208 |
4.930 |
0.000 |
| z |
0.000 |
0.000 |
3.283 |
<r2> (average value of r
2) Å
2
| <r2> |
110.532 |
| (<r2>)1/2 |
10.513 |