Jump to
S1C2
Vibrational Frequencies calculated at M06-2X/6-31+G**
Geometric Data calculated at M06-2X/6-31+G**
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at M06-2X/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -323.507539 |
| Energy at 298.15K | -323.516736 |
| HF Energy | -323.507539 |
| Nuclear repulsion energy | 244.000311 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3152 |
3002 |
19.62 |
|
|
|
| 2 |
A |
3149 |
2998 |
24.73 |
|
|
|
| 3 |
A |
3142 |
2992 |
28.44 |
|
|
|
| 4 |
A |
3112 |
2963 |
12.21 |
|
|
|
| 5 |
A |
3087 |
2939 |
20.91 |
|
|
|
| 6 |
A |
3066 |
2920 |
26.00 |
|
|
|
| 7 |
A |
3063 |
2916 |
12.21 |
|
|
|
| 8 |
A |
1824 |
1737 |
244.59 |
|
|
|
| 9 |
A |
1522 |
1449 |
11.95 |
|
|
|
| 10 |
A |
1509 |
1437 |
9.17 |
|
|
|
| 11 |
A |
1497 |
1426 |
3.22 |
|
|
|
| 12 |
A |
1486 |
1415 |
2.68 |
|
|
|
| 13 |
A |
1431 |
1362 |
7.95 |
|
|
|
| 14 |
A |
1417 |
1349 |
9.97 |
|
|
|
| 15 |
A |
1381 |
1315 |
0.42 |
|
|
|
| 16 |
A |
1314 |
1251 |
21.05 |
|
|
|
| 17 |
A |
1297 |
1235 |
4.83 |
|
|
|
| 18 |
A |
1193 |
1136 |
13.38 |
|
|
|
| 19 |
A |
1134 |
1080 |
52.95 |
|
|
|
| 20 |
A |
1116 |
1063 |
18.75 |
|
|
|
| 21 |
A |
1026 |
977 |
8.08 |
|
|
|
| 22 |
A |
957 |
912 |
331.51 |
|
|
|
| 23 |
A |
899 |
856 |
92.15 |
|
|
|
| 24 |
A |
886 |
844 |
59.96 |
|
|
|
| 25 |
A |
797 |
759 |
2.96 |
|
|
|
| 26 |
A |
658 |
627 |
4.37 |
|
|
|
| 27 |
A |
467 |
445 |
4.50 |
|
|
|
| 28 |
A |
394 |
375 |
2.60 |
|
|
|
| 29 |
A |
295 |
281 |
0.59 |
|
|
|
| 30 |
A |
276 |
263 |
0.38 |
|
|
|
| 31 |
A |
194 |
185 |
0.50 |
|
|
|
| 32 |
A |
109 |
104 |
0.91 |
|
|
|
| 33 |
A |
72 |
68 |
0.07 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23460.8 cm
-1
Scaled (by 0.9522) Zero Point Vibrational Energy (zpe) 22339.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/6-31+G**
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.173 |
-0.921 |
0.183 |
| C2 |
-1.751 |
0.458 |
-0.319 |
| C3 |
-0.404 |
0.898 |
0.223 |
| O4 |
0.574 |
-0.032 |
-0.278 |
| N5 |
1.785 |
0.208 |
0.311 |
| O6 |
2.556 |
-0.556 |
-0.147 |
| H7 |
-3.154 |
-1.195 |
-0.213 |
| H8 |
-2.235 |
-0.936 |
1.276 |
| H9 |
-1.455 |
-1.684 |
-0.127 |
| H10 |
-1.709 |
0.470 |
-1.414 |
| H11 |
-2.485 |
1.214 |
-0.019 |
| H12 |
-0.137 |
1.903 |
-0.117 |
| H13 |
-0.383 |
0.869 |
1.318 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5276 | 2.5375 | 2.9235 | 4.1178 | 4.7543 | 1.0929 | 1.0950 | 1.0927 | 2.1680 | 2.1678 | 3.4943 | 2.7746 |
C2 | 1.5276 | | 1.5169 | 2.3757 | 3.6001 | 4.4276 | 2.1713 | 2.1735 | 2.1713 | 1.0954 | 1.0960 | 2.1753 | 2.1730 | C3 | 2.5375 | 1.5169 | | 1.4390 | 2.2970 | 3.3183 | 3.4832 | 2.7970 | 2.8094 | 2.1372 | 2.1188 | 1.0943 | 1.0955 | O4 | 2.9235 | 2.3757 | 1.4390 | | 1.3677 | 2.0544 | 3.9054 | 3.3348 | 2.6207 | 2.5990 | 3.3128 | 2.0673 | 2.0673 | N5 | 4.1178 | 3.6001 | 2.2970 | 1.3677 | | 1.1775 | 5.1609 | 4.2898 | 3.7772 | 3.9055 | 4.3995 | 2.5983 | 2.4807 | O6 | 4.7543 | 4.4276 | 3.3183 | 2.0544 | 1.1775 | | 5.7457 | 5.0124 | 4.1664 | 4.5658 | 5.3441 | 3.6466 | 3.5799 | H7 | 1.0929 | 2.1713 | 3.4832 | 3.9054 | 5.1609 | 5.7457 | | 1.7691 | 1.7701 | 2.5103 | 2.5084 | 4.3254 | 3.7797 | H8 | 1.0950 | 2.1735 | 2.7970 | 3.3348 | 4.2898 | 5.0124 | 1.7691 | | 1.7712 | 3.0805 | 2.5229 | 3.7950 | 2.5867 | H9 | 1.0927 | 2.1713 | 2.8094 | 2.6207 | 3.7772 | 4.1664 | 1.7701 | 1.7712 | | 2.5222 | 3.0782 | 3.8215 | 3.1238 | H10 | 2.1680 | 1.0954 | 2.1372 | 2.5990 | 3.9055 | 4.5658 | 2.5103 | 3.0805 | 2.5222 | | 1.7611 | 2.4916 | 3.0634 | H11 | 2.1678 | 1.0960 | 2.1188 | 3.3128 | 4.3995 | 5.3441 | 2.5084 | 2.5229 | 3.0782 | 1.7611 | | 2.4487 | 2.5154 | H12 | 3.4943 | 2.1753 | 1.0943 | 2.0673 | 2.5983 | 3.6466 | 4.3254 | 3.7950 | 3.8215 | 2.4916 | 2.4487 | | 1.7857 | H13 | 2.7746 | 2.1730 | 1.0955 | 2.0673 | 2.4807 | 3.5799 | 3.7797 | 2.5867 | 3.1238 | 3.0634 | 2.5154 | 1.7857 | |
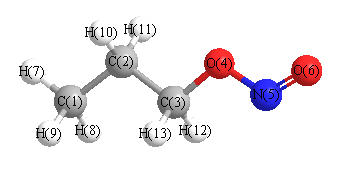 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
112.911 |
|
C1 |
C2 |
H10 |
110.407 |
| C1 |
C2 |
H11 |
110.350 |
|
C2 |
C1 |
H7 |
110.814 |
| C2 |
C1 |
H8 |
110.871 |
|
C2 |
C1 |
H9 |
110.830 |
| C2 |
C3 |
O4 |
106.946 |
|
C2 |
C3 |
H12 |
111.807 |
| C2 |
C3 |
H13 |
111.546 |
|
C3 |
C2 |
H10 |
108.719 |
| C3 |
C2 |
H11 |
107.272 |
|
C3 |
O4 |
N5 |
109.824 |
| O4 |
C3 |
H12 |
108.618 |
|
O4 |
C3 |
H13 |
108.545 |
| O4 |
N5 |
O6 |
107.403 |
|
H7 |
C1 |
H8 |
107.915 |
| H7 |
C1 |
H9 |
108.173 |
|
H8 |
C1 |
H9 |
108.122 |
| H10 |
C2 |
H11 |
106.957 |
|
H12 |
C3 |
H13 |
109.262 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.503 |
|
|
|
| 2 |
C |
-0.276 |
|
|
|
| 3 |
C |
-0.119 |
|
|
|
| 4 |
O |
-0.100 |
|
|
|
| 5 |
N |
-0.155 |
|
|
|
| 6 |
O |
-0.020 |
|
|
|
| 7 |
H |
0.166 |
|
|
|
| 8 |
H |
0.162 |
|
|
|
| 9 |
H |
0.172 |
|
|
|
| 10 |
H |
0.175 |
|
|
|
| 11 |
H |
0.162 |
|
|
|
| 12 |
H |
0.167 |
|
|
|
| 13 |
H |
0.167 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.770 |
-0.914 |
0.826 |
3.031 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-39.388 |
0.356 |
-0.885 |
| y |
0.356 |
-35.422 |
-1.217 |
| z |
-0.885 |
-1.217 |
-37.444 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.955 |
0.356 |
-0.885 |
| y |
0.356 |
2.993 |
-1.217 |
| z |
-0.885 |
-1.217 |
-0.038 |
|
| Polar |
|---|
| 3z2-r2 | -0.077 |
|---|
| x2-y2 | -3.965 |
|---|
| xy | 0.356 |
|---|
| xz | -0.885 |
|---|
| yz | -1.217 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.426 |
-1.035 |
0.069 |
| y |
-1.035 |
7.491 |
0.006 |
| z |
0.069 |
0.006 |
6.581 |
<r2> (average value of r
2) Å
2
| <r2> |
187.483 |
| (<r2>)1/2 |
13.692 |