Jump to
S1C2
Energy calculated at M06-2X/6-311G*
| | hartrees |
|---|
| Energy at 0K | -499.417770 |
| Energy at 298.15K | |
| HF Energy | -499.417770 |
| Nuclear repulsion energy | 45.253789 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3205 |
3205 |
9.34 |
102.09 |
0.14 |
0.25 |
| 2 |
A' |
1439 |
1439 |
10.38 |
2.64 |
0.74 |
0.85 |
| 3 |
A' |
847 |
847 |
46.00 |
6.09 |
0.34 |
0.51 |
| 4 |
A' |
356 |
356 |
74.05 |
0.72 |
0.01 |
0.03 |
| 5 |
A" |
3356 |
3356 |
0.20 |
50.43 |
0.75 |
0.86 |
| 6 |
A" |
1027 |
1027 |
0.92 |
4.41 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 5114.8 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 5114.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/6-311G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.013 |
1.124 |
0.000 |
| Cl2 |
-0.013 |
-0.586 |
0.000 |
| H3 |
0.146 |
1.609 |
0.949 |
| H4 |
0.146 |
1.609 |
-0.949 |
Atom - Atom Distances (Å)
| |
C1 |
Cl2 |
H3 |
H4 |
| C1 | | 1.7095 | 1.0774 | 1.0774 |
Cl2 | 1.7095 | | 2.3966 | 2.3966 | H3 | 1.0774 | 2.3966 | | 1.8971 | H4 | 1.0774 | 2.3966 | 1.8971 | |
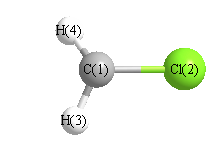 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Br2 |
C1 |
H3 |
116.789 |
|
Br2 |
C1 |
H4 |
116.789 |
| H3 |
C1 |
H4 |
123.378 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.529 |
|
|
|
| 2 |
Cl |
-0.015 |
|
|
|
| 3 |
H |
0.272 |
|
|
|
| 4 |
H |
0.272 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.306 |
1.300 |
0.000 |
1.335 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-20.834 |
0.505 |
0.000 |
| y |
0.505 |
-17.598 |
0.000 |
| z |
0.000 |
0.000 |
-18.729 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.670 |
0.505 |
0.000 |
| y |
0.505 |
2.183 |
0.000 |
| z |
0.000 |
0.000 |
0.486 |
|
| Polar |
|---|
| 3z2-r2 | 0.973 |
|---|
| x2-y2 | -3.235 |
|---|
| xy | 0.505 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.694 |
0.054 |
0.000 |
| y |
0.054 |
4.507 |
0.000 |
| z |
0.000 |
0.000 |
2.276 |
<r2> (average value of r
2) Å
2
| <r2> |
32.337 |
| (<r2>)1/2 |
5.687 |
Jump to
S1C1
Energy calculated at M06-2X/6-311G*
| | hartrees |
|---|
| Energy at 0K | -499.417685 |
| Energy at 298.15K | |
| HF Energy | -499.417685 |
| Nuclear repulsion energy | 45.297266 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3215 |
3215 |
7.27 |
100.79 |
0.15 |
0.26 |
| 2 |
A1 |
1436 |
1436 |
11.06 |
|
|
|
| 3 |
A1 |
851 |
851 |
43.67 |
|
|
|
| 4 |
B1 |
253i |
253i |
90.44 |
|
|
|
| 5 |
B2 |
3371 |
3371 |
0.00 |
|
|
|
| 6 |
B2 |
1016 |
1016 |
0.95 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4818.6 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 4818.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/6-311G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.120 |
| Cl2 |
0.000 |
0.000 |
0.586 |
| H3 |
0.000 |
0.954 |
-1.619 |
| H4 |
0.000 |
-0.954 |
-1.619 |
Atom - Atom Distances (Å)
| |
C1 |
Cl2 |
H3 |
H4 |
| C1 | | 1.7064 | 1.0764 | 1.0764 |
Cl2 | 1.7064 | | 2.4028 | 2.4028 | H3 | 1.0764 | 2.4028 | | 1.9074 | H4 | 1.0764 | 2.4028 | 1.9074 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Br2 |
C1 |
H3 |
117.622 |
|
Br2 |
C1 |
H4 |
117.622 |
| H3 |
C1 |
H4 |
124.756 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.538 |
|
|
|
| 2 |
Cl |
-0.011 |
|
|
|
| 3 |
H |
0.274 |
|
|
|
| 4 |
H |
0.274 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-1.304 |
1.304 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-20.902 |
0.000 |
0.000 |
| y |
0.000 |
-18.671 |
0.000 |
| z |
0.000 |
0.000 |
-17.505 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.814 |
0.000 |
0.000 |
| y |
0.000 |
0.532 |
0.000 |
| z |
0.000 |
0.000 |
2.282 |
|
| Polar |
|---|
| 3z2-r2 | 4.564 |
|---|
| x2-y2 | -2.231 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.678 |
0.000 |
0.000 |
| y |
0.000 |
2.268 |
0.000 |
| z |
0.000 |
0.000 |
4.499 |
<r2> (average value of r
2) Å
2
| <r2> |
32.316 |
| (<r2>)1/2 |
5.685 |