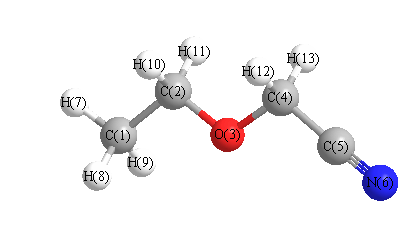
Vibrational Frequencies calculated at M06-2X/3-21G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3199 |
3030 |
11.72 |
|
|
|
| 2 |
A' |
3116 |
2951 |
6.26 |
|
|
|
| 3 |
A' |
3081 |
2918 |
29.41 |
|
|
|
| 4 |
A' |
3060 |
2899 |
12.26 |
|
|
|
| 5 |
A' |
2399 |
2272 |
0.39 |
|
|
|
| 6 |
A' |
1604 |
1519 |
4.87 |
|
|
|
| 7 |
A' |
1577 |
1493 |
1.76 |
|
|
|
| 8 |
A' |
1572 |
1489 |
10.65 |
|
|
|
| 9 |
A' |
1484 |
1406 |
6.10 |
|
|
|
| 10 |
A' |
1471 |
1394 |
11.13 |
|
|
|
| 11 |
A' |
1409 |
1334 |
45.82 |
|
|
|
| 12 |
A' |
1181 |
1119 |
79.45 |
|
|
|
| 13 |
A' |
1140 |
1080 |
77.31 |
|
|
|
| 14 |
A' |
1037 |
983 |
6.14 |
|
|
|
| 15 |
A' |
971 |
920 |
16.55 |
|
|
|
| 16 |
A' |
912 |
864 |
3.32 |
|
|
|
| 17 |
A' |
600 |
569 |
0.67 |
|
|
|
| 18 |
A' |
420 |
398 |
1.67 |
|
|
|
| 19 |
A' |
312 |
296 |
1.88 |
|
|
|
| 20 |
A' |
135 |
127 |
2.12 |
|
|
|
| 21 |
A" |
3190 |
3022 |
13.87 |
|
|
|
| 22 |
A" |
3113 |
2949 |
33.12 |
|
|
|
| 23 |
A" |
3090 |
2927 |
18.56 |
|
|
|
| 24 |
A" |
1559 |
1477 |
8.37 |
|
|
|
| 25 |
A" |
1342 |
1271 |
0.42 |
|
|
|
| 26 |
A" |
1295 |
1227 |
1.21 |
|
|
|
| 27 |
A" |
1215 |
1151 |
9.46 |
|
|
|
| 28 |
A" |
1080 |
1023 |
0.85 |
|
|
|
| 29 |
A" |
877 |
831 |
1.96 |
|
|
|
| 30 |
A" |
456 |
432 |
0.10 |
|
|
|
| 31 |
A" |
281 |
267 |
0.69 |
|
|
|
| 32 |
A" |
132 |
125 |
9.08 |
|
|
|
| 33 |
A" |
114 |
108 |
0.08 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24212.6 cm
-1
Scaled (by 0.9472) Zero Point Vibrational Energy (zpe) 22934.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.589 |
|
|
|
| 2 |
C |
-0.147 |
|
|
|
| 3 |
O |
-0.498 |
|
|
|
| 4 |
C |
-0.132 |
|
|
|
| 5 |
C |
0.305 |
|
|
|
| 6 |
N |
-0.445 |
|
|
|
| 7 |
H |
0.199 |
|
|
|
| 8 |
H |
0.223 |
|
|
|
| 9 |
H |
0.223 |
|
|
|
| 10 |
H |
0.192 |
|
|
|
| 11 |
H |
0.192 |
|
|
|
| 12 |
H |
0.239 |
|
|
|
| 13 |
H |
0.239 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-4.548 |
1.203 |
0.000 |
4.705 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.059 |
5.626 |
0.000 |
| y |
5.626 |
-37.889 |
0.000 |
| z |
0.000 |
0.000 |
-35.655 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-5.287 |
5.626 |
0.000 |
| y |
5.626 |
0.968 |
0.000 |
| z |
0.000 |
0.000 |
4.319 |
|
| Polar |
|---|
| 3z2-r2 | 8.638 |
|---|
| x2-y2 | -4.170 |
|---|
| xy | 5.626 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.402 |
-1.677 |
0.000 |
| y |
-1.677 |
7.426 |
0.000 |
| z |
0.000 |
0.000 |
5.098 |
<r2> (average value of r
2) Å
2
| <r2> |
231.820 |
| (<r2>)1/2 |
15.226 |