Jump to
S1C2
Energy calculated at M06-2X/6-31G
| | hartrees |
|---|
| Energy at 0K | -551.166966 |
| Energy at 298.15K | |
| HF Energy | -551.166966 |
| Nuclear repulsion energy | 349.151820 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/6-31G
Geometric Data calculated at M06-2X/6-31G
Point Group is D4h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
1.016 |
0.000 |
| C2 |
1.016 |
0.000 |
0.000 |
| C3 |
0.000 |
-1.016 |
0.000 |
| C4 |
-1.016 |
0.000 |
0.000 |
| F5 |
0.000 |
2.360 |
0.000 |
| F6 |
2.360 |
0.000 |
0.000 |
| F7 |
0.000 |
-2.360 |
0.000 |
| F8 |
-2.360 |
0.000 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.4373 | 2.0326 | 1.4373 | 1.3439 | 2.5697 | 3.3766 | 2.5697 |
C2 | 1.4373 | | 1.4373 | 2.0326 | 2.5697 | 1.3439 | 2.5697 | 3.3766 | C3 | 2.0326 | 1.4373 | | 1.4373 | 3.3766 | 2.5697 | 1.3439 | 2.5697 | C4 | 1.4373 | 2.0326 | 1.4373 | | 2.5697 | 3.3766 | 2.5697 | 1.3439 | F5 | 1.3439 | 2.5697 | 3.3766 | 2.5697 | | 3.3379 | 4.7205 | 3.3379 | F6 | 2.5697 | 1.3439 | 2.5697 | 3.3766 | 3.3379 | | 3.3379 | 4.7205 | F7 | 3.3766 | 2.5697 | 1.3439 | 2.5697 | 4.7205 | 3.3379 | | 3.3379 | F8 | 2.5697 | 3.3766 | 2.5697 | 1.3439 | 3.3379 | 4.7205 | 3.3379 | |
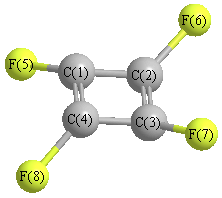 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
135.000 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
135.000 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
135.000 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
135.000 |
| C3 |
C2 |
F6 |
135.000 |
|
C3 |
C4 |
F8 |
135.000 |
| C4 |
C1 |
F5 |
135.000 |
|
C4 |
C3 |
F7 |
135.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.414 |
|
|
|
| 2 |
C |
0.134 |
|
|
|
| 3 |
C |
0.414 |
|
|
|
| 4 |
C |
0.134 |
|
|
|
| 5 |
F |
-0.240 |
|
|
|
| 6 |
F |
-0.307 |
|
|
|
| 7 |
F |
-0.240 |
|
|
|
| 8 |
F |
-0.307 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-51.279 |
0.000 |
0.000 |
| y |
0.000 |
-40.558 |
0.000 |
| z |
0.000 |
0.000 |
-40.632 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-10.685 |
0.000 |
0.000 |
| y |
0.000 |
5.398 |
0.000 |
| z |
0.000 |
0.000 |
5.287 |
|
| Polar |
|---|
| 3z2-r2 | 10.574 |
|---|
| x2-y2 | -10.721 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.711 |
0.000 |
0.000 |
| y |
0.000 |
7.599 |
0.000 |
| z |
0.000 |
0.000 |
2.488 |
<r2> (average value of r
2) Å
2
| <r2> |
252.914 |
| (<r2>)1/2 |
15.903 |
Jump to
S1C1
Energy calculated at M06-2X/6-31G
| | hartrees |
|---|
| Energy at 0K | -551.253443 |
| Energy at 298.15K | -551.253126 |
| HF Energy | -551.253443 |
| Nuclear repulsion energy | 347.702449 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at M06-2X/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
1937 |
1937 |
0.00 |
|
|
|
| 2 |
Ag |
1261 |
1261 |
0.00 |
|
|
|
| 3 |
Ag |
625 |
625 |
0.00 |
|
|
|
| 4 |
Ag |
219 |
219 |
0.00 |
|
|
|
| 5 |
Ag |
101 |
101 |
0.00 |
|
|
|
| 6 |
Au |
1339 |
1339 |
310.54 |
|
|
|
| 7 |
Au |
897 |
897 |
33.41 |
|
|
|
| 8 |
Au |
582 |
582 |
2.25 |
|
|
|
| 9 |
Au |
211 |
211 |
0.91 |
|
|
|
| 10 |
Au |
159 |
159 |
0.00 |
|
|
|
| 11 |
Bg |
1410 |
1410 |
0.00 |
|
|
|
| 12 |
Bg |
738 |
738 |
0.00 |
|
|
|
| 13 |
Bg |
485 |
485 |
0.00 |
|
|
|
| 14 |
Bg |
476 |
476 |
0.00 |
|
|
|
| 15 |
Bu |
1925 |
1925 |
52.36 |
|
|
|
| 16 |
Bu |
975 |
975 |
145.91 |
|
|
|
| 17 |
Bu |
290 |
290 |
9.91 |
|
|
|
| 18 |
Bu |
201 |
201 |
2.36 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 6915.8 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 6915.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at M06-2X/6-31G
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.025 |
0.792 |
0.661 |
| C2 |
-0.025 |
-0.792 |
0.661 |
| C3 |
-0.025 |
-0.792 |
-0.661 |
| C4 |
0.025 |
0.792 |
-0.661 |
| F5 |
-0.025 |
1.683 |
1.669 |
| F6 |
0.025 |
-1.683 |
1.669 |
| F7 |
0.025 |
-1.683 |
-1.669 |
| F8 |
-0.025 |
1.683 |
-1.669 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.5852 | 2.0643 | 1.3224 | 1.3456 | 2.6723 | 3.3992 | 2.4948 |
C2 | 1.5852 | | 1.3224 | 2.0643 | 2.6723 | 1.3456 | 2.4948 | 3.3992 | C3 | 2.0643 | 1.3224 | | 1.5852 | 3.3992 | 2.4948 | 1.3456 | 2.6723 | C4 | 1.3224 | 2.0643 | 1.5852 | | 2.4948 | 3.3992 | 2.6723 | 1.3456 | F5 | 1.3456 | 2.6723 | 3.3992 | 2.4948 | | 3.3663 | 4.7401 | 3.3371 | F6 | 2.6723 | 1.3456 | 2.4948 | 3.3992 | 3.3663 | | 3.3371 | 4.7401 | F7 | 3.3992 | 2.4948 | 1.3456 | 2.6723 | 4.7401 | 3.3371 | | 3.3663 | F8 | 2.4948 | 3.3992 | 2.6723 | 1.3456 | 3.3371 | 4.7401 | 3.3663 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
131.339 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
138.471 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
131.339 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
138.471 |
| C3 |
C2 |
F6 |
138.471 |
|
C3 |
C4 |
F8 |
131.339 |
| C4 |
C1 |
F5 |
138.471 |
|
C4 |
C3 |
F7 |
131.339 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at M06-2X/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.283 |
|
|
|
| 2 |
C |
0.283 |
|
|
|
| 3 |
C |
0.283 |
|
|
|
| 4 |
C |
0.283 |
|
|
|
| 5 |
F |
-0.283 |
|
|
|
| 6 |
F |
-0.283 |
|
|
|
| 7 |
F |
-0.283 |
|
|
|
| 8 |
F |
-0.283 |
|
|
|
Electric dipole moments
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.208 |
0.059 |
0.000 |
| y |
0.059 |
-45.886 |
0.000 |
| z |
0.000 |
0.000 |
-46.262 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.866 |
0.059 |
0.000 |
| y |
0.059 |
-2.651 |
0.000 |
| z |
0.000 |
0.000 |
-3.215 |
|
| Polar |
|---|
| 3z2-r2 | -6.429 |
|---|
| x2-y2 | 5.678 |
|---|
| xy | 0.059 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.390 |
-0.025 |
0.000 |
| y |
-0.025 |
5.121 |
0.000 |
| z |
0.000 |
0.000 |
7.237 |
<r2> (average value of r
2) Å
2
| <r2> |
255.344 |
| (<r2>)1/2 |
15.979 |