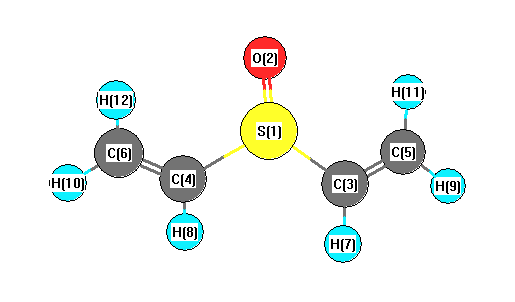
Vibrational Frequencies calculated at CCD/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3238 |
3107 |
0.92 |
|
|
|
| 2 |
A' |
3209 |
3079 |
6.50 |
|
|
|
| 3 |
A' |
3140 |
3012 |
5.14 |
|
|
|
| 4 |
A' |
1678 |
1610 |
5.66 |
|
|
|
| 5 |
A' |
1446 |
1388 |
18.04 |
|
|
|
| 6 |
A' |
1309 |
1256 |
6.22 |
|
|
|
| 7 |
A' |
1049 |
1007 |
2.44 |
|
|
|
| 8 |
A' |
1021 |
979 |
76.40 |
|
|
|
| 9 |
A' |
962 |
923 |
1.70 |
|
|
|
| 10 |
A' |
773 |
742 |
14.10 |
|
|
|
| 11 |
A' |
693 |
665 |
64.79 |
|
|
|
| 12 |
A' |
599 |
575 |
0.13 |
|
|
|
| 13 |
A' |
457 |
438 |
1.96 |
|
|
|
| 14 |
A' |
278 |
267 |
6.16 |
|
|
|
| 15 |
A' |
198 |
190 |
5.63 |
|
|
|
| 16 |
A' |
84 |
81 |
0.83 |
|
|
|
| 17 |
A" |
3238 |
3107 |
2.87 |
|
|
|
| 18 |
A" |
3206 |
3077 |
3.34 |
|
|
|
| 19 |
A" |
3139 |
3012 |
6.28 |
|
|
|
| 20 |
A" |
1670 |
1602 |
16.49 |
|
|
|
| 21 |
A" |
1443 |
1385 |
1.16 |
|
|
|
| 22 |
A" |
1291 |
1239 |
25.64 |
|
|
|
| 23 |
A" |
1038 |
996 |
1.69 |
|
|
|
| 24 |
A" |
1016 |
975 |
56.05 |
|
|
|
| 25 |
A" |
960 |
921 |
3.63 |
|
|
|
| 26 |
A" |
615 |
590 |
9.85 |
|
|
|
| 27 |
A" |
587 |
563 |
4.84 |
|
|
|
| 28 |
A" |
438 |
420 |
3.51 |
|
|
|
| 29 |
A" |
239 |
230 |
16.60 |
|
|
|
| 30 |
A" |
141 |
135 |
0.45 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19576.9 cm
-1
Scaled (by 0.9595) Zero Point Vibrational Energy (zpe) 18784.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.