Jump to
S1C2
Vibrational Frequencies calculated at wB97X-D/6-31G*
Geometric Data calculated at wB97X-D/6-31G*
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at wB97X-D/6-31G*
| | hartrees |
|---|
| Energy at 0K | -260.948771 |
| Energy at 298.15K | -260.953830 |
| HF Energy | -260.948771 |
| Nuclear repulsion energy | 127.599514 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3587 |
3402 |
38.05 |
|
|
|
| 2 |
A' |
1652 |
1567 |
82.93 |
|
|
|
| 3 |
A' |
1464 |
1389 |
229.94 |
|
|
|
| 4 |
A' |
1059 |
1005 |
18.66 |
|
|
|
| 5 |
A' |
826 |
784 |
157.64 |
|
|
|
| 6 |
A' |
742 |
703 |
102.31 |
|
|
|
| 7 |
A' |
657 |
623 |
112.11 |
|
|
|
| 8 |
A" |
3724 |
3532 |
53.95 |
|
|
|
| 9 |
A" |
1775 |
1683 |
314.41 |
|
|
|
| 10 |
A" |
1276 |
1210 |
45.15 |
|
|
|
| 11 |
A" |
583 |
553 |
2.11 |
|
|
|
| 12 |
A" |
435 |
413 |
29.91 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8890.0 cm
-1
Scaled (by 0.9485) Zero Point Vibrational Energy (zpe) 8432.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.073 |
-1.241 |
0.000 |
| N2 |
0.006 |
0.142 |
0.000 |
| O3 |
0.006 |
0.681 |
1.091 |
| O4 |
0.006 |
0.681 |
-1.091 |
| H5 |
-0.322 |
-1.604 |
-0.859 |
| H6 |
-0.322 |
-1.604 |
0.859 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
O3 |
O4 |
H5 |
H6 |
| N1 | | 1.3842 | 2.2109 | 2.2109 | 1.0126 | 1.0126 |
N2 | 1.3842 | | 1.2169 | 1.2169 | 1.9728 | 1.9728 | O3 | 2.2109 | 1.2169 | | 2.1817 | 3.0214 | 2.3199 | O4 | 2.2109 | 1.2169 | 2.1817 | | 2.3199 | 3.0214 | H5 | 1.0126 | 1.9728 | 3.0214 | 2.3199 | | 1.7176 | H6 | 1.0126 | 1.9728 | 2.3199 | 3.0214 | 1.7176 | |
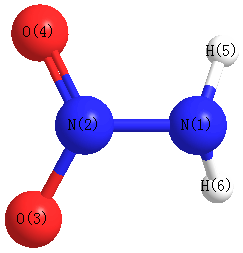 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
N2 |
O3 |
116.271 |
|
N1 |
N2 |
O4 |
116.271 |
| N2 |
N1 |
H5 |
109.828 |
|
N2 |
N1 |
H6 |
109.828 |
| O3 |
N2 |
O4 |
127.393 |
|
H5 |
N1 |
H6 |
116.022 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.545 |
|
|
|
| 2 |
N |
0.601 |
|
|
|
| 3 |
O |
-0.404 |
|
|
|
| 4 |
O |
-0.404 |
|
|
|
| 5 |
H |
0.376 |
|
|
|
| 6 |
H |
0.376 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.358 |
-3.642 |
0.000 |
3.887 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-21.888 |
2.441 |
0.000 |
| y |
2.441 |
-19.977 |
0.000 |
| z |
0.000 |
0.000 |
-23.793 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.003 |
2.441 |
0.000 |
| y |
2.441 |
2.863 |
0.000 |
| z |
0.000 |
0.000 |
-2.860 |
|
| Polar |
|---|
| 3z2-r2 | -5.721 |
|---|
| x2-y2 | -1.910 |
|---|
| xy | 2.441 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.580 |
0.075 |
0.000 |
| y |
0.075 |
3.850 |
0.000 |
| z |
0.000 |
0.000 |
4.248 |
<r2> (average value of r
2) Å
2
| <r2> |
57.913 |
| (<r2>)1/2 |
7.610 |