Jump to
S1C2
Energy calculated at wB97X-D/SDD
| | hartrees |
|---|
| Energy at 0K | -187.949872 |
| Energy at 298.15K | -187.953824 |
| HF Energy | -187.949872 |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/SDD
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3800 |
3800 |
26.91 |
|
|
|
| 2 |
A |
3669 |
3669 |
0.00 |
|
|
|
| 3 |
A |
2393 |
2393 |
0.00 |
|
|
|
| 4 |
A |
1691 |
1691 |
0.00 |
|
|
|
| 5 |
A |
1162 |
1162 |
6.01 |
|
|
|
| 6 |
A |
845 |
845 |
0.00 |
|
|
|
| 7 |
A |
531 |
531 |
0.00 |
|
|
|
| 8 |
A |
477 |
477 |
202.99 |
|
|
|
| 9 |
A |
426 |
426 |
233.68 |
|
|
|
| 10 |
A |
197 |
197 |
30.09 |
|
|
|
| 11 |
B |
3800 |
3800 |
26.79 |
|
|
|
| 12 |
B |
3674 |
3674 |
24.58 |
|
|
|
| 13 |
B |
1698 |
1698 |
84.78 |
|
|
|
| 14 |
B |
1445 |
1445 |
151.37 |
|
|
|
| 15 |
B |
1162 |
1162 |
5.97 |
|
|
|
| 16 |
B |
477 |
477 |
203.47 |
|
|
|
| 17 |
B |
426 |
426 |
234.97 |
|
|
|
| 18 |
B |
197 |
197 |
29.96 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14034.0 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 14034.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/SDD
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.008 |
0.610 |
-0.008 |
| C2 |
-0.008 |
-0.610 |
-0.008 |
| N3 |
-0.008 |
1.980 |
-0.032 |
| N4 |
0.008 |
-1.980 |
-0.032 |
| H5 |
-0.427 |
2.474 |
0.749 |
| H6 |
0.764 |
2.464 |
-0.476 |
| H7 |
0.427 |
-2.474 |
0.749 |
| H8 |
-0.764 |
-2.464 |
-0.476 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
N4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.2208 | 1.3704 | 2.5908 | 2.0577 | 2.0556 | 3.2029 | 3.2040 |
C2 | 1.2208 | | 2.5908 | 1.3704 | 3.2029 | 3.2040 | 2.0577 | 2.0556 | N3 | 1.3704 | 2.5908 | | 3.9609 | 1.0137 | 1.0138 | 4.5427 | 4.5298 | N4 | 2.5908 | 1.3704 | 3.9609 | | 4.5427 | 4.5298 | 1.0137 | 1.0138 | H5 | 2.0577 | 3.2029 | 1.0137 | 4.5427 | | 1.7082 | 5.0202 | 5.0980 | H6 | 2.0556 | 3.2040 | 1.0138 | 4.5298 | 1.7082 | | 5.0980 | 5.1593 | H7 | 3.2029 | 2.0577 | 4.5427 | 1.0137 | 5.0202 | 5.0980 | | 1.7082 | H8 | 3.2040 | 2.0556 | 4.5298 | 1.0138 | 5.0980 | 5.1593 | 1.7082 | |
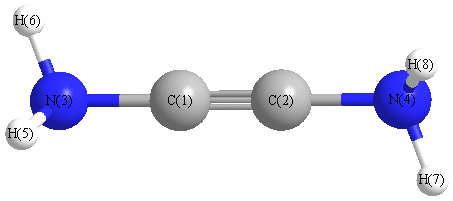 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N4 |
178.224 |
|
C1 |
N3 |
H5 |
118.567 |
| C1 |
N3 |
H6 |
118.356 |
|
C2 |
C1 |
N3 |
178.224 |
| C2 |
N4 |
H7 |
118.567 |
|
C2 |
N4 |
H8 |
118.356 |
| H5 |
N3 |
H6 |
114.820 |
|
H7 |
N4 |
H8 |
114.820 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/SDD
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.031 |
|
|
|
| 2 |
C |
0.031 |
|
|
|
| 3 |
N |
-0.696 |
|
|
|
| 4 |
N |
-0.696 |
|
|
|
| 5 |
H |
0.333 |
|
|
|
| 6 |
H |
0.333 |
|
|
|
| 7 |
H |
0.333 |
|
|
|
| 8 |
H |
0.333 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.007 |
0.007 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.904 |
-0.022 |
0.000 |
| y |
-0.022 |
-10.354 |
0.000 |
| z |
0.000 |
0.000 |
-23.887 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.783 |
-0.022 |
0.000 |
| y |
-0.022 |
13.541 |
0.000 |
| z |
0.000 |
0.000 |
-6.758 |
|
| Polar |
|---|
| 3z2-r2 | -13.516 |
|---|
| x2-y2 | -13.550 |
|---|
| xy | -0.022 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.595 |
-0.002 |
0.000 |
| y |
-0.002 |
9.003 |
0.000 |
| z |
0.000 |
0.000 |
2.596 |
<r2> (average value of r
2) Å
2
| <r2> |
98.146 |
| (<r2>)1/2 |
9.907 |
Jump to
S1C1
Energy calculated at wB97X-D/SDD
| | hartrees |
|---|
| Energy at 0K | -187.949852 |
| Energy at 298.15K | -187.953780 |
| HF Energy | -187.949852 |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/SDD
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3669 |
3669 |
0.00 |
|
|
|
| 2 |
A1 |
2393 |
2393 |
0.00 |
|
|
|
| 3 |
A1 |
1687 |
1687 |
0.00 |
|
|
|
| 4 |
A1 |
845 |
845 |
0.00 |
|
|
|
| 5 |
B1 |
527 |
527 |
0.00 |
|
|
|
| 6 |
B2 |
3674 |
3674 |
24.80 |
|
|
|
| 7 |
B2 |
1694 |
1694 |
85.54 |
|
|
|
| 8 |
B2 |
1445 |
1445 |
151.01 |
|
|
|
| 9 |
E |
3800 |
3800 |
26.99 |
|
|
|
| 9 |
E |
3800 |
3800 |
26.99 |
|
|
|
| 10 |
E |
1159 |
1159 |
5.89 |
|
|
|
| 10 |
E |
1159 |
1159 |
5.89 |
|
|
|
| 11 |
E |
475 |
475 |
179.74 |
|
|
|
| 11 |
E |
475 |
475 |
179.74 |
|
|
|
| 12 |
E |
421 |
421 |
257.35 |
|
|
|
| 12 |
E |
421 |
421 |
257.35 |
|
|
|
| 13 |
E |
194 |
194 |
30.41 |
|
|
|
| 13 |
E |
194 |
194 |
30.41 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 14016.2 cm
-1
Scaled (by 1) Zero Point Vibrational Energy (zpe) 14016.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/SDD
Point Group is D2d
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.612 |
| C2 |
0.000 |
0.000 |
-0.612 |
| N3 |
0.000 |
0.000 |
1.963 |
| N4 |
0.000 |
0.000 |
-1.963 |
| H5 |
0.000 |
0.863 |
2.481 |
| H6 |
0.000 |
-0.863 |
2.481 |
| H7 |
0.863 |
0.000 |
-2.481 |
| H8 |
-0.863 |
0.000 |
-2.481 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
N4 |
H5 |
H6 |
H7 |
H8 |
| C1 | | 1.2240 | 1.3508 | 2.5748 | 2.0587 | 2.0587 | 3.2113 | 3.2113 |
C2 | 1.2240 | | 2.5748 | 1.3508 | 3.2113 | 3.2113 | 2.0587 | 2.0587 | N3 | 1.3508 | 2.5748 | | 3.9256 | 1.0065 | 1.0065 | 4.5270 | 4.5270 | N4 | 2.5748 | 1.3508 | 3.9256 | | 4.5270 | 4.5270 | 1.0065 | 1.0065 | H5 | 2.0587 | 3.2113 | 1.0065 | 4.5270 | | 1.7252 | 5.1103 | 5.1103 | H6 | 2.0587 | 3.2113 | 1.0065 | 4.5270 | 1.7252 | | 5.1103 | 5.1103 | H7 | 3.2113 | 2.0587 | 4.5270 | 1.0065 | 5.1103 | 5.1103 | | 1.7252 | H8 | 3.2113 | 2.0587 | 4.5270 | 1.0065 | 5.1103 | 5.1103 | 1.7252 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N4 |
180.000 |
|
C1 |
N3 |
H5 |
121.011 |
| C1 |
N3 |
H6 |
121.011 |
|
C2 |
C1 |
N3 |
180.000 |
| C2 |
N4 |
H7 |
121.011 |
|
C2 |
N4 |
H8 |
121.011 |
| H5 |
N3 |
H6 |
117.978 |
|
H7 |
N4 |
H8 |
117.978 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/SDD
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.031 |
|
|
|
| 2 |
C |
0.031 |
|
|
|
| 3 |
N |
-0.696 |
|
|
|
| 4 |
N |
-0.696 |
|
|
|
| 5 |
H |
0.333 |
|
|
|
| 6 |
H |
0.333 |
|
|
|
| 7 |
H |
0.333 |
|
|
|
| 8 |
H |
0.333 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-23.893 |
0.000 |
0.000 |
| y |
0.000 |
-23.893 |
0.000 |
| z |
0.000 |
0.000 |
-10.369 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-6.762 |
0.000 |
0.000 |
| y |
0.000 |
-6.762 |
0.000 |
| z |
0.000 |
0.000 |
13.524 |
|
| Polar |
|---|
| 3z2-r2 | 27.048 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.595 |
0.000 |
0.000 |
| y |
0.000 |
2.595 |
0.000 |
| z |
0.000 |
0.000 |
8.998 |
<r2> (average value of r
2) Å
2
| <r2> |
98.141 |
| (<r2>)1/2 |
9.907 |