Jump to
S1C2
S1C3
Energy calculated at wB97X-D/6-311G**
| | hartrees |
|---|
| Energy at 0K | -306.458908 |
| Energy at 298.15K | |
| HF Energy | -306.458908 |
| Nuclear repulsion energy | 226.547677 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3179 |
3040 |
0.00 |
|
|
|
| 2 |
Ag |
3061 |
2928 |
0.00 |
|
|
|
| 3 |
Ag |
1850 |
1770 |
0.00 |
|
|
|
| 4 |
Ag |
1457 |
1394 |
0.00 |
|
|
|
| 5 |
Ag |
1406 |
1345 |
0.00 |
|
|
|
| 6 |
Ag |
1297 |
1240 |
0.00 |
|
|
|
| 7 |
Ag |
1019 |
975 |
0.00 |
|
|
|
| 8 |
Ag |
697 |
667 |
0.00 |
|
|
|
| 9 |
Ag |
532 |
509 |
0.00 |
|
|
|
| 10 |
Ag |
366 |
350 |
0.00 |
|
|
|
| 11 |
Au |
3131 |
2995 |
6.43 |
|
|
|
| 12 |
Au |
1457 |
1394 |
25.44 |
|
|
|
| 13 |
Au |
962 |
920 |
6.11 |
|
|
|
| 14 |
Au |
355 |
339 |
4.65 |
|
|
|
| 15 |
Au |
79 |
75 |
6.75 |
|
|
|
| 16 |
Au |
61i |
58i |
4.52 |
|
|
|
| 17 |
Bg |
3131 |
2995 |
0.00 |
|
|
|
| 18 |
Bg |
1462 |
1399 |
0.00 |
|
|
|
| 19 |
Bg |
1076 |
1029 |
0.00 |
|
|
|
| 20 |
Bg |
628 |
600 |
0.00 |
|
|
|
| 21 |
Bg |
36i |
34i |
0.00 |
|
|
|
| 22 |
Bu |
3179 |
3041 |
13.56 |
|
|
|
| 23 |
Bu |
3061 |
2928 |
1.07 |
|
|
|
| 24 |
Bu |
1844 |
1764 |
268.52 |
|
|
|
| 25 |
Bu |
1459 |
1395 |
30.02 |
|
|
|
| 26 |
Bu |
1393 |
1332 |
80.74 |
|
|
|
| 27 |
Bu |
1143 |
1094 |
85.80 |
|
|
|
| 28 |
Bu |
922 |
882 |
19.76 |
|
|
|
| 29 |
Bu |
552 |
528 |
42.31 |
|
|
|
| 30 |
Bu |
242 |
232 |
16.77 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20421.7 cm
-1
Scaled (by 0.9565) Zero Point Vibrational Energy (zpe) 19533.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-311G**
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.054 |
0.744 |
0.000 |
| C2 |
0.054 |
-0.744 |
0.000 |
| C3 |
1.177 |
1.582 |
0.000 |
| C4 |
-1.177 |
-1.582 |
0.000 |
| O5 |
-1.177 |
1.228 |
0.000 |
| O6 |
1.177 |
-1.228 |
0.000 |
| H7 |
0.894 |
2.632 |
0.000 |
| H8 |
-0.894 |
-2.632 |
0.000 |
| H9 |
1.783 |
1.357 |
0.880 |
| H10 |
1.783 |
1.357 |
-0.880 |
| H11 |
-1.783 |
-1.357 |
0.880 |
| H12 |
-1.783 |
-1.357 |
-0.880 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
O5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| C1 | | 1.4928 | 1.4881 | 2.5830 | 1.2225 | 2.3245 | 2.1121 | 3.4794 | 2.1267 | 2.1267 | 2.8602 | 2.8602 |
C2 | 1.4928 | | 2.5830 | 1.4881 | 2.3245 | 1.2225 | 3.4794 | 2.1121 | 2.8602 | 2.8602 | 2.1267 | 2.1267 | C3 | 1.4881 | 2.5830 | | 3.9427 | 2.3795 | 2.8096 | 1.0874 | 4.6950 | 1.0920 | 1.0920 | 4.2626 | 4.2626 | C4 | 2.5830 | 1.4881 | 3.9427 | | 2.8096 | 2.3795 | 4.6950 | 1.0874 | 4.2626 | 4.2626 | 1.0920 | 1.0920 | O5 | 1.2225 | 2.3245 | 2.3795 | 2.8096 | | 3.4011 | 2.5019 | 3.8700 | 3.0904 | 3.0904 | 2.7969 | 2.7969 | O6 | 2.3245 | 1.2225 | 2.8096 | 2.3795 | 3.4011 | | 3.8700 | 2.5019 | 2.7969 | 2.7969 | 3.0904 | 3.0904 | H7 | 2.1121 | 3.4794 | 1.0874 | 4.6950 | 2.5019 | 3.8700 | | 5.5593 | 1.7860 | 1.7860 | 4.8840 | 4.8840 | H8 | 3.4794 | 2.1121 | 4.6950 | 1.0874 | 3.8700 | 2.5019 | 5.5593 | | 4.8840 | 4.8840 | 1.7860 | 1.7860 | H9 | 2.1267 | 2.8602 | 1.0920 | 4.2626 | 3.0904 | 2.7969 | 1.7860 | 4.8840 | | 1.7593 | 4.4815 | 4.8145 | H10 | 2.1267 | 2.8602 | 1.0920 | 4.2626 | 3.0904 | 2.7969 | 1.7860 | 4.8840 | 1.7593 | | 4.8145 | 4.4815 | H11 | 2.8602 | 2.1267 | 4.2626 | 1.0920 | 2.7969 | 3.0904 | 4.8840 | 1.7860 | 4.4815 | 4.8145 | | 1.7593 | H12 | 2.8602 | 2.1267 | 4.2626 | 1.0920 | 2.7969 | 3.0904 | 4.8840 | 1.7860 | 4.8145 | 4.4815 | 1.7593 | |
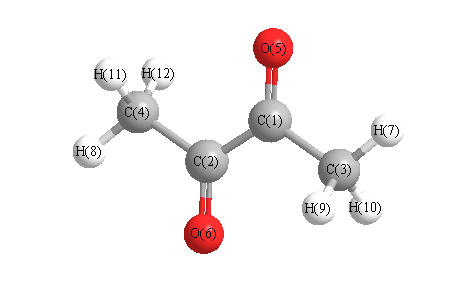 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
120.120 |
|
C1 |
C2 |
O6 |
117.414 |
| C1 |
C3 |
H7 |
109.197 |
|
C1 |
C3 |
H9 |
110.086 |
| C1 |
C3 |
H10 |
110.086 |
|
C2 |
C1 |
C3 |
120.120 |
| C2 |
C1 |
O5 |
117.414 |
|
C2 |
C4 |
H8 |
109.197 |
| C2 |
C4 |
H11 |
110.086 |
|
C2 |
C4 |
H12 |
110.086 |
| C3 |
C1 |
O5 |
122.466 |
|
C4 |
C2 |
O6 |
122.466 |
| H7 |
C3 |
H9 |
110.066 |
|
H7 |
C3 |
H10 |
110.066 |
| H8 |
C4 |
H11 |
110.066 |
|
H8 |
C4 |
H12 |
110.066 |
| H9 |
C3 |
H10 |
107.323 |
|
H11 |
C4 |
H12 |
107.323 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.148 |
|
|
|
| 2 |
C |
0.148 |
|
|
|
| 3 |
C |
-0.322 |
|
|
|
| 4 |
C |
-0.322 |
|
|
|
| 5 |
O |
-0.275 |
|
|
|
| 6 |
O |
-0.275 |
|
|
|
| 7 |
H |
0.147 |
|
|
|
| 8 |
H |
0.147 |
|
|
|
| 9 |
H |
0.151 |
|
|
|
| 10 |
H |
0.151 |
|
|
|
| 11 |
H |
0.151 |
|
|
|
| 12 |
H |
0.151 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.405 |
7.361 |
0.000 |
| y |
7.361 |
-36.506 |
0.000 |
| z |
0.000 |
0.000 |
-33.448 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.428 |
7.361 |
0.000 |
| y |
7.361 |
-0.579 |
0.000 |
| z |
0.000 |
0.000 |
4.007 |
|
| Polar |
|---|
| 3z2-r2 | 8.014 |
|---|
| x2-y2 | -1.899 |
|---|
| xy | 7.361 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.298 |
0.146 |
0.000 |
| y |
0.146 |
7.571 |
0.000 |
| z |
0.000 |
0.000 |
5.039 |
<r2> (average value of r
2) Å
2
| <r2> |
162.320 |
| (<r2>)1/2 |
12.740 |
Jump to
S1C1
S1C3
Energy calculated at wB97X-D/6-311G**
| | hartrees |
|---|
| Energy at 0K | -306.458918 |
| Energy at 298.15K | -306.465055 |
| HF Energy | -306.458918 |
| Nuclear repulsion energy | 226.544441 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3184 |
3046 |
0.00 |
|
|
|
| 2 |
A |
3139 |
3003 |
6.42 |
|
|
|
| 3 |
A |
3068 |
2934 |
0.17 |
|
|
|
| 4 |
A |
1850 |
1770 |
0.17 |
|
|
|
| 5 |
A |
1467 |
1403 |
24.25 |
|
|
|
| 6 |
A |
1464 |
1400 |
0.55 |
|
|
|
| 7 |
A |
1411 |
1350 |
0.54 |
|
|
|
| 8 |
A |
1298 |
1241 |
0.02 |
|
|
|
| 9 |
A |
1024 |
980 |
0.01 |
|
|
|
| 10 |
A |
970 |
928 |
6.15 |
|
|
|
| 11 |
A |
697 |
667 |
0.01 |
|
|
|
| 12 |
A |
533 |
510 |
0.02 |
|
|
|
| 13 |
A |
374 |
358 |
0.63 |
|
|
|
| 14 |
A |
358 |
342 |
4.12 |
|
|
|
| 15 |
A |
116 |
111 |
0.13 |
|
|
|
| 16 |
A |
60 |
57 |
11.09 |
|
|
|
| 17 |
B |
3185 |
3046 |
12.90 |
|
|
|
| 18 |
B |
3139 |
3003 |
0.51 |
|
|
|
| 19 |
B |
3068 |
2934 |
1.11 |
|
|
|
| 20 |
B |
1844 |
1764 |
267.71 |
|
|
|
| 21 |
B |
1472 |
1408 |
1.19 |
|
|
|
| 22 |
B |
1465 |
1401 |
29.97 |
|
|
|
| 23 |
B |
1399 |
1338 |
78.97 |
|
|
|
| 24 |
B |
1145 |
1095 |
84.26 |
|
|
|
| 25 |
B |
1081 |
1034 |
0.26 |
|
|
|
| 26 |
B |
927 |
887 |
21.14 |
|
|
|
| 27 |
B |
635 |
607 |
0.16 |
|
|
|
| 28 |
B |
553 |
528 |
42.08 |
|
|
|
| 29 |
B |
251 |
240 |
16.43 |
|
|
|
| 30 |
B |
112 |
107 |
0.26 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20643.0 cm
-1
Scaled (by 0.9565) Zero Point Vibrational Energy (zpe) 19745.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-311G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.076 |
0.773 |
-0.027 |
| C2 |
0.076 |
-0.773 |
-0.027 |
| C3 |
1.183 |
1.583 |
0.062 |
| C4 |
-1.183 |
-1.583 |
0.062 |
| O5 |
-1.183 |
1.250 |
-0.069 |
| O6 |
1.183 |
-1.250 |
-0.069 |
| H7 |
1.063 |
2.525 |
-0.473 |
| H8 |
-1.063 |
-2.525 |
-0.473 |
| H9 |
1.364 |
1.816 |
1.117 |
| H10 |
2.032 |
1.010 |
-0.305 |
| H11 |
-1.364 |
-1.816 |
1.117 |
| H12 |
-2.032 |
-1.010 |
-0.305 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
O5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| C1 | | 1.5532 | 1.4997 | 2.6043 | 1.2064 | 2.3829 | 2.1367 | 3.4710 | 2.1145 | 2.1396 | 3.1098 | 2.6616 |
C2 | 1.5532 | | 2.6043 | 1.4997 | 2.3829 | 1.2064 | 3.4710 | 2.1367 | 3.1098 | 2.6616 | 2.1145 | 2.1396 | C3 | 1.4997 | 2.6043 | | 3.9521 | 2.3934 | 2.8354 | 1.0901 | 4.7120 | 1.0956 | 1.0878 | 4.3765 | 4.1469 | C4 | 2.6043 | 1.4997 | 3.9521 | | 2.8354 | 2.3934 | 4.7120 | 1.0901 | 4.3765 | 4.1469 | 1.0956 | 1.0878 | O5 | 1.2064 | 2.3829 | 2.3934 | 2.8354 | | 3.4421 | 2.6146 | 3.7979 | 2.8665 | 3.2331 | 3.2919 | 2.4256 | O6 | 2.3829 | 1.2064 | 2.8354 | 2.3934 | 3.4421 | | 3.7979 | 2.6146 | 3.2919 | 2.4256 | 2.8665 | 3.2331 | H7 | 2.1367 | 3.4710 | 1.0901 | 4.7120 | 2.6146 | 3.7979 | | 5.4788 | 1.7669 | 1.8059 | 5.2214 | 4.7016 | H8 | 3.4710 | 2.1367 | 4.7120 | 1.0901 | 3.7979 | 2.6146 | 5.4788 | | 5.2214 | 4.7016 | 1.7669 | 1.8059 | H9 | 2.1145 | 3.1098 | 1.0956 | 4.3765 | 2.8665 | 3.2919 | 1.7669 | 5.2214 | | 1.7658 | 4.5428 | 4.6418 | H10 | 2.1396 | 2.6616 | 1.0878 | 4.1469 | 3.2331 | 2.4256 | 1.8059 | 4.7016 | 1.7658 | | 4.6418 | 4.5389 | H11 | 3.1098 | 2.1145 | 4.3765 | 1.0956 | 3.2919 | 2.8665 | 5.2214 | 1.7669 | 4.5428 | 4.6418 | | 1.7658 | H12 | 2.6616 | 2.1396 | 4.1469 | 1.0878 | 2.4256 | 3.2331 | 4.7016 | 1.8059 | 4.6418 | 4.5389 | 1.7658 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
117.085 |
|
C1 |
C2 |
O6 |
118.885 |
| C1 |
C3 |
H7 |
110.184 |
|
C1 |
C3 |
H9 |
108.116 |
| C1 |
C3 |
H10 |
110.555 |
|
C2 |
C1 |
C3 |
117.085 |
| C2 |
C1 |
O5 |
118.885 |
|
C2 |
C4 |
H8 |
110.184 |
| C2 |
C4 |
H11 |
108.116 |
|
C2 |
C4 |
H12 |
110.555 |
| C3 |
C1 |
O5 |
124.009 |
|
C4 |
C2 |
O6 |
124.009 |
| H7 |
C3 |
H9 |
107.880 |
|
H7 |
C3 |
H10 |
112.023 |
| H8 |
C4 |
H11 |
107.880 |
|
H8 |
C4 |
H12 |
112.023 |
| H9 |
C3 |
H10 |
107.941 |
|
H11 |
C4 |
H12 |
107.941 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.148 |
|
|
|
| 2 |
C |
0.148 |
|
|
|
| 3 |
C |
-0.322 |
|
|
|
| 4 |
C |
-0.322 |
|
|
|
| 5 |
O |
-0.276 |
|
|
|
| 6 |
O |
-0.276 |
|
|
|
| 7 |
H |
0.147 |
|
|
|
| 8 |
H |
0.147 |
|
|
|
| 9 |
H |
0.151 |
|
|
|
| 10 |
H |
0.152 |
|
|
|
| 11 |
H |
0.151 |
|
|
|
| 12 |
H |
0.152 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.132 |
0.132 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.413 |
7.365 |
0.000 |
| y |
7.365 |
-36.488 |
0.000 |
| z |
0.000 |
0.000 |
-33.446 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.446 |
7.365 |
0.000 |
| y |
7.365 |
-0.558 |
0.000 |
| z |
0.000 |
0.000 |
4.004 |
|
| Polar |
|---|
| 3z2-r2 | 8.008 |
|---|
| x2-y2 | -1.925 |
|---|
| xy | 7.365 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.288 |
0.151 |
0.000 |
| y |
0.151 |
7.575 |
0.000 |
| z |
0.000 |
0.000 |
5.042 |
<r2> (average value of r
2) Å
2
| <r2> |
162.341 |
| (<r2>)1/2 |
12.741 |
Jump to
S1C1
S1C2
Energy calculated at wB97X-D/6-311G**
| | hartrees |
|---|
| Energy at 0K | -306.458864 |
| Energy at 298.15K | |
| HF Energy | -306.458864 |
| Nuclear repulsion energy | 226.556116 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3182 |
3044 |
0.00 |
|
|
|
| 2 |
Ag |
3134 |
2998 |
0.00 |
|
|
|
| 3 |
Ag |
3064 |
2931 |
0.00 |
|
|
|
| 4 |
Ag |
1849 |
1769 |
0.00 |
|
|
|
| 5 |
Ag |
1461 |
1398 |
0.00 |
|
|
|
| 6 |
Ag |
1460 |
1396 |
0.00 |
|
|
|
| 7 |
Ag |
1403 |
1342 |
0.00 |
|
|
|
| 8 |
Ag |
1298 |
1241 |
0.00 |
|
|
|
| 9 |
Ag |
1073 |
1026 |
0.00 |
|
|
|
| 10 |
Ag |
1019 |
974 |
0.00 |
|
|
|
| 11 |
Ag |
697 |
666 |
0.00 |
|
|
|
| 12 |
Ag |
624 |
597 |
0.00 |
|
|
|
| 13 |
Ag |
532 |
509 |
0.00 |
|
|
|
| 14 |
Ag |
365 |
349 |
0.00 |
|
|
|
| 15 |
Ag |
47 |
45 |
0.00 |
|
|
|
| 16 |
Au |
3183 |
3044 |
13.51 |
|
|
|
| 17 |
Au |
3134 |
2998 |
6.48 |
|
|
|
| 18 |
Au |
3064 |
2931 |
1.10 |
|
|
|
| 19 |
Au |
1844 |
1764 |
268.45 |
|
|
|
| 20 |
Au |
1461 |
1398 |
30.69 |
|
|
|
| 21 |
Au |
1456 |
1393 |
25.30 |
|
|
|
| 22 |
Au |
1390 |
1330 |
79.07 |
|
|
|
| 23 |
Au |
1144 |
1095 |
86.50 |
|
|
|
| 24 |
Au |
958 |
916 |
6.08 |
|
|
|
| 25 |
Au |
922 |
882 |
19.74 |
|
|
|
| 26 |
Au |
551 |
527 |
42.77 |
|
|
|
| 27 |
Au |
348 |
333 |
5.14 |
|
|
|
| 28 |
Au |
240 |
229 |
16.67 |
|
|
|
| 29 |
Au |
76 |
72 |
3.11 |
|
|
|
| 30 |
Au |
33i |
32i |
7.62 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20472.7 cm
-1
Scaled (by 0.9565) Zero Point Vibrational Energy (zpe) 19582.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-311G**
Point Group is Ci
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.011 |
0.000 |
-0.775 |
| C2 |
-0.011 |
-0.000 |
0.775 |
| C3 |
1.359 |
-0.000 |
-1.427 |
| C4 |
-1.359 |
0.000 |
1.427 |
| O5 |
-1.035 |
-0.000 |
-1.377 |
| O6 |
1.035 |
0.000 |
1.377 |
| H7 |
1.933 |
0.870 |
-1.097 |
| H8 |
-1.933 |
-0.870 |
1.097 |
| H9 |
1.933 |
-0.870 |
-1.097 |
| H10 |
1.254 |
-0.000 |
-2.510 |
| H11 |
-1.933 |
0.870 |
1.097 |
| H12 |
-1.254 |
0.000 |
2.510 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
O5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
| C1 | | 1.5501 | 1.4974 | 2.5933 | 1.2067 | 2.3827 | 2.1341 | 2.8358 | 2.1341 | 2.1340 | 2.8355 | 3.5199 |
C2 | 1.5501 | | 2.5933 | 1.4974 | 2.3827 | 1.2067 | 2.8358 | 2.1341 | 2.8355 | 3.5199 | 2.1341 | 2.1340 | C3 | 1.4974 | 2.5933 | | 3.9410 | 2.3947 | 2.8220 | 1.0933 | 4.2385 | 1.0933 | 1.0882 | 4.2384 | 4.7248 | C4 | 2.5933 | 1.4974 | 3.9410 | | 2.8220 | 2.3947 | 4.2385 | 1.0933 | 4.2384 | 4.7248 | 1.0933 | 1.0882 | O5 | 1.2067 | 2.3827 | 2.3947 | 2.8220 | | 3.4445 | 3.1053 | 2.7719 | 3.1054 | 2.5539 | 2.7716 | 3.8925 | O6 | 2.3827 | 1.2067 | 2.8220 | 2.3947 | 3.4445 | | 2.7719 | 3.1053 | 2.7716 | 3.8925 | 3.1054 | 2.5539 | H7 | 2.1341 | 2.8358 | 1.0933 | 4.2385 | 3.1053 | 2.7719 | | 4.7738 | 1.7408 | 1.7929 | 4.4449 | 4.8910 | H8 | 2.8358 | 2.1341 | 4.2385 | 1.0933 | 2.7719 | 3.1053 | 4.7738 | | 4.4449 | 4.8910 | 1.7408 | 1.7929 | H9 | 2.1341 | 2.8355 | 1.0933 | 4.2384 | 3.1054 | 2.7716 | 1.7408 | 4.4449 | | 1.7929 | 4.7735 | 4.8909 | H10 | 2.1340 | 3.5199 | 1.0882 | 4.7248 | 2.5539 | 3.8925 | 1.7929 | 4.8910 | 1.7929 | | 4.8909 | 5.6110 | H11 | 2.8355 | 2.1341 | 4.2384 | 1.0933 | 2.7716 | 3.1054 | 4.4449 | 1.7408 | 4.7735 | 4.8909 | | 1.7929 | H12 | 3.5199 | 2.1340 | 4.7248 | 1.0882 | 3.8925 | 2.5539 | 4.8910 | 1.7929 | 4.8909 | 5.6110 | 1.7929 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C4 |
116.623 |
|
C1 |
C2 |
O6 |
119.084 |
| C1 |
C3 |
H7 |
109.946 |
|
C1 |
C3 |
H9 |
109.944 |
| C1 |
C3 |
H10 |
110.242 |
|
C2 |
C1 |
C3 |
116.623 |
| C2 |
C1 |
O5 |
119.084 |
|
C2 |
C4 |
H8 |
109.946 |
| C2 |
C4 |
H11 |
109.944 |
|
C2 |
C4 |
H12 |
110.242 |
| C3 |
C1 |
O5 |
124.293 |
|
C4 |
C2 |
O6 |
124.293 |
| H7 |
C3 |
H9 |
105.525 |
|
H7 |
C3 |
H10 |
110.544 |
| H8 |
C4 |
H11 |
105.525 |
|
H8 |
C4 |
H12 |
110.544 |
| H9 |
C3 |
H10 |
110.543 |
|
H11 |
C4 |
H12 |
110.543 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.148 |
|
|
|
| 2 |
C |
0.148 |
|
|
|
| 3 |
C |
-0.322 |
|
|
|
| 4 |
C |
-0.322 |
|
|
|
| 5 |
O |
-0.275 |
|
|
|
| 6 |
O |
-0.275 |
|
|
|
| 7 |
H |
0.151 |
|
|
|
| 8 |
H |
0.151 |
|
|
|
| 9 |
H |
0.151 |
|
|
|
| 10 |
H |
0.147 |
|
|
|
| 11 |
H |
0.151 |
|
|
|
| 12 |
H |
0.147 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.672 |
-0.000 |
-7.379 |
| y |
-0.000 |
-33.446 |
-0.000 |
| z |
-7.379 |
-0.000 |
-38.237 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.830 |
-0.000 |
-7.379 |
| y |
-0.000 |
4.008 |
-0.000 |
| z |
-7.379 |
-0.000 |
-3.178 |
|
| Polar |
|---|
| 3z2-r2 | -6.356 |
|---|
| x2-y2 | -3.226 |
|---|
| xy | -0.000 |
|---|
| xz | -7.379 |
|---|
| yz | -0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.316 |
0.000 |
-0.051 |
| y |
0.000 |
5.038 |
0.000 |
| z |
-0.051 |
0.000 |
7.537 |
<r2> (average value of r
2) Å
2
| <r2> |
162.317 |
| (<r2>)1/2 |
12.740 |