Jump to
S1C2
S1C3
Energy calculated at wB97X-D/6-311G**
| | hartrees |
|---|
| Energy at 0K | -268.330590 |
| Energy at 298.15K | |
| HF Energy | -268.330590 |
| Nuclear repulsion energy | 195.068639 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3134 |
2998 |
24.79 |
|
|
|
| 2 |
A |
3023 |
2891 |
0.88 |
|
|
|
| 3 |
A |
3013 |
2882 |
140.73 |
|
|
|
| 4 |
A |
1556 |
1488 |
2.42 |
|
|
|
| 5 |
A |
1524 |
1458 |
0.66 |
|
|
|
| 6 |
A |
1395 |
1334 |
6.39 |
|
|
|
| 7 |
A |
1267 |
1212 |
12.01 |
|
|
|
| 8 |
A |
1236 |
1183 |
72.65 |
|
|
|
| 9 |
A |
1183 |
1131 |
136.16 |
|
|
|
| 10 |
A |
1151 |
1101 |
32.03 |
|
|
|
| 11 |
A |
993 |
950 |
3.49 |
|
|
|
| 12 |
A |
962 |
920 |
3.87 |
|
|
|
| 13 |
A |
747 |
715 |
0.55 |
|
|
|
| 14 |
A |
263 |
252 |
0.58 |
|
|
|
| 15 |
B |
3139 |
3003 |
37.78 |
|
|
|
| 16 |
B |
3057 |
2924 |
77.77 |
|
|
|
| 17 |
B |
3030 |
2898 |
84.71 |
|
|
|
| 18 |
B |
1518 |
1452 |
4.17 |
|
|
|
| 19 |
B |
1448 |
1385 |
8.00 |
|
|
|
| 20 |
B |
1354 |
1295 |
0.22 |
|
|
|
| 21 |
B |
1248 |
1194 |
4.84 |
|
|
|
| 22 |
B |
1160 |
1109 |
20.57 |
|
|
|
| 23 |
B |
1104 |
1056 |
35.25 |
|
|
|
| 24 |
B |
983 |
941 |
71.63 |
|
|
|
| 25 |
B |
916 |
877 |
19.72 |
|
|
|
| 26 |
B |
651 |
622 |
4.25 |
|
|
|
| 27 |
B |
99i |
94i |
18.31 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20478.2 cm
-1
Scaled (by 0.9565) Zero Point Vibrational Energy (zpe) 19587.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-311G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
1.201 |
| C2 |
-0.298 |
0.697 |
-0.942 |
| C3 |
0.298 |
-0.697 |
-0.942 |
| O4 |
0.000 |
1.143 |
0.372 |
| O5 |
0.000 |
-1.143 |
0.372 |
| H6 |
0.900 |
0.042 |
1.826 |
| H7 |
-0.900 |
-0.042 |
1.826 |
| H8 |
-1.383 |
0.662 |
-1.107 |
| H9 |
1.383 |
-0.662 |
-1.107 |
| H10 |
-0.161 |
-1.386 |
-1.650 |
| H11 |
0.161 |
1.386 |
-1.650 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 2.2731 | 2.2731 | 1.4116 | 1.4116 | 1.0962 | 1.0962 | 2.7706 | 2.7706 | 3.1744 | 3.1744 |
C2 | 2.2731 | | 1.5159 | 1.4193 | 2.2800 | 3.0860 | 2.9266 | 1.0973 | 2.1678 | 2.2039 | 1.0900 | C3 | 2.2731 | 1.5159 | | 2.2800 | 1.4193 | 2.9266 | 3.0860 | 2.1678 | 1.0973 | 1.0900 | 2.2039 | O4 | 1.4116 | 1.4193 | 2.2800 | | 2.2850 | 2.0333 | 2.0795 | 2.0808 | 2.7119 | 3.2416 | 2.0432 | O5 | 1.4116 | 2.2800 | 1.4193 | 2.2850 | | 2.0795 | 2.0333 | 2.7119 | 2.0808 | 2.0432 | 3.2416 | H6 | 1.0962 | 3.0860 | 2.9266 | 2.0333 | 2.0795 | | 1.8022 | 3.7674 | 3.0536 | 3.9044 | 3.7990 | H7 | 1.0962 | 2.9266 | 3.0860 | 2.0795 | 2.0333 | 1.8022 | | 3.0536 | 3.7674 | 3.7990 | 3.9044 | H8 | 2.7706 | 1.0973 | 2.1678 | 2.0808 | 2.7119 | 3.7674 | 3.0536 | | 3.0660 | 2.4453 | 1.7900 | H9 | 2.7706 | 2.1678 | 1.0973 | 2.7119 | 2.0808 | 3.0536 | 3.7674 | 3.0660 | | 1.7900 | 2.4453 | H10 | 3.1744 | 2.2039 | 1.0900 | 3.2416 | 2.0432 | 3.9044 | 3.7990 | 2.4453 | 1.7900 | | 2.7903 | H11 | 3.1744 | 1.0900 | 2.2039 | 2.0432 | 3.2416 | 3.7990 | 3.9044 | 1.7900 | 2.4453 | 2.7903 | |
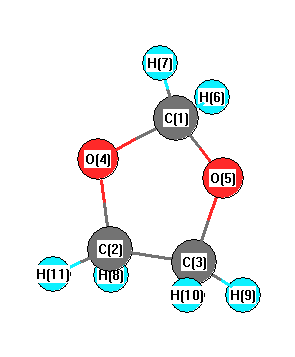 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O4 |
C2 |
106.829 |
|
C1 |
O5 |
C3 |
106.829 |
| C2 |
C3 |
O5 |
101.883 |
|
C2 |
C3 |
H9 |
111.094 |
| C2 |
C3 |
H10 |
114.516 |
|
C3 |
C2 |
O4 |
101.883 |
| C3 |
C2 |
H8 |
111.094 |
|
C3 |
C2 |
H11 |
114.516 |
| O4 |
C1 |
O5 |
108.066 |
|
O4 |
C1 |
H6 |
107.683 |
| O4 |
C1 |
H7 |
111.418 |
|
O4 |
C2 |
H8 |
110.904 |
| O4 |
C2 |
H11 |
108.320 |
|
O5 |
C1 |
H6 |
111.418 |
| O5 |
C1 |
H7 |
107.683 |
|
O5 |
C3 |
H9 |
110.904 |
| O5 |
C3 |
H10 |
108.320 |
|
H6 |
C1 |
H7 |
110.574 |
| H8 |
C2 |
H11 |
109.844 |
|
H9 |
C3 |
H10 |
109.844 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.180 |
|
|
|
| 2 |
C |
-0.057 |
|
|
|
| 3 |
C |
-0.057 |
|
|
|
| 4 |
O |
-0.376 |
|
|
|
| 5 |
O |
-0.376 |
|
|
|
| 6 |
H |
0.104 |
|
|
|
| 7 |
H |
0.104 |
|
|
|
| 8 |
H |
0.114 |
|
|
|
| 9 |
H |
0.114 |
|
|
|
| 10 |
H |
0.125 |
|
|
|
| 11 |
H |
0.125 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-1.216 |
1.216 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.071 |
-0.265 |
0.000 |
| y |
-0.265 |
-34.674 |
0.000 |
| z |
0.000 |
0.000 |
-25.635 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.083 |
-0.265 |
0.000 |
| y |
-0.265 |
-7.321 |
0.000 |
| z |
0.000 |
0.000 |
6.237 |
|
| Polar |
|---|
| 3z2-r2 | 12.475 |
|---|
| x2-y2 | 5.603 |
|---|
| xy | -0.265 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.224 |
-0.095 |
0.000 |
| y |
-0.095 |
5.110 |
0.000 |
| z |
0.000 |
0.000 |
6.520 |
<r2> (average value of r
2) Å
2
| <r2> |
92.698 |
| (<r2>)1/2 |
9.628 |
Jump to
S1C1
S1C3
Energy calculated at wB97X-D/6-311G**
| | hartrees |
|---|
| Energy at 0K | -268.331198 |
| Energy at 298.15K | -268.339582 |
| HF Energy | -268.331198 |
| Nuclear repulsion energy | 195.089809 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3143 |
3006 |
30.17 |
|
|
|
| 2 |
A |
3137 |
3001 |
42.20 |
|
|
|
| 3 |
A |
3105 |
2970 |
34.89 |
|
|
|
| 4 |
A |
3057 |
2924 |
64.12 |
|
|
|
| 5 |
A |
3018 |
2887 |
54.50 |
|
|
|
| 6 |
A |
2951 |
2822 |
120.76 |
|
|
|
| 7 |
A |
1556 |
1489 |
2.41 |
|
|
|
| 8 |
A |
1538 |
1471 |
1.08 |
|
|
|
| 9 |
A |
1527 |
1461 |
3.32 |
|
|
|
| 10 |
A |
1442 |
1379 |
10.60 |
|
|
|
| 11 |
A |
1389 |
1329 |
0.54 |
|
|
|
| 12 |
A |
1359 |
1300 |
0.16 |
|
|
|
| 13 |
A |
1290 |
1234 |
6.90 |
|
|
|
| 14 |
A |
1244 |
1189 |
3.25 |
|
|
|
| 15 |
A |
1232 |
1178 |
2.78 |
|
|
|
| 16 |
A |
1208 |
1156 |
104.83 |
|
|
|
| 17 |
A |
1169 |
1118 |
14.83 |
|
|
|
| 18 |
A |
1139 |
1089 |
107.45 |
|
|
|
| 19 |
A |
1102 |
1054 |
41.28 |
|
|
|
| 20 |
A |
1015 |
971 |
33.71 |
|
|
|
| 21 |
A |
979 |
937 |
64.78 |
|
|
|
| 22 |
A |
959 |
918 |
21.12 |
|
|
|
| 23 |
A |
877 |
839 |
8.54 |
|
|
|
| 24 |
A |
755 |
722 |
1.05 |
|
|
|
| 25 |
A |
705 |
674 |
5.72 |
|
|
|
| 26 |
A |
299 |
286 |
12.32 |
|
|
|
| 27 |
A |
66 |
64 |
3.62 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20630.7 cm
-1
Scaled (by 0.9565) Zero Point Vibrational Energy (zpe) 19733.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-311G**
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.191 |
-0.055 |
0.078 |
| C2 |
-1.005 |
-0.676 |
-0.135 |
| C3 |
-0.870 |
0.803 |
0.198 |
| O4 |
0.314 |
-1.164 |
0.058 |
| O5 |
0.440 |
1.079 |
-0.254 |
| H6 |
1.621 |
0.035 |
1.089 |
| H7 |
1.988 |
-0.172 |
-0.661 |
| H8 |
-1.314 |
-0.818 |
-1.178 |
| H9 |
-0.954 |
0.974 |
1.281 |
| H10 |
-1.572 |
1.451 |
-0.327 |
| H11 |
-1.692 |
-1.214 |
0.521 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 2.2915 | 2.2355 | 1.4140 | 1.3996 | 1.1018 | 1.0936 | 2.9044 | 2.6653 | 3.1721 | 3.1379 |
C2 | 2.2915 | | 1.5215 | 1.4193 | 2.2760 | 2.9828 | 3.0798 | 1.0970 | 2.1748 | 2.2093 | 1.0917 | C3 | 2.2355 | 1.5215 | | 2.3001 | 1.4134 | 2.7548 | 3.1393 | 2.1718 | 1.1001 | 1.0895 | 2.2013 | O4 | 1.4140 | 1.4193 | 2.3001 | | 2.2679 | 2.0516 | 2.0745 | 2.0731 | 2.7702 | 3.2468 | 2.0589 | O5 | 1.3996 | 2.2760 | 1.4134 | 2.2679 | | 2.0703 | 2.0307 | 2.7440 | 2.0765 | 2.0474 | 3.2251 | H6 | 1.1018 | 2.9828 | 2.7548 | 2.0516 | 2.0703 | | 1.7999 | 3.8053 | 2.7471 | 3.7683 | 3.5853 | H7 | 1.0936 | 3.0798 | 3.1393 | 2.0745 | 2.0307 | 1.7999 | | 3.4040 | 3.7064 | 3.9261 | 4.0026 | H8 | 2.9044 | 1.0970 | 2.1718 | 2.0731 | 2.7440 | 3.8053 | 3.4040 | | 3.0641 | 2.4372 | 1.7848 | H9 | 2.6653 | 2.1748 | 1.1001 | 2.7702 | 2.0765 | 2.7471 | 3.7064 | 3.0641 | | 1.7875 | 2.4306 | H10 | 3.1721 | 2.2093 | 1.0895 | 3.2468 | 2.0474 | 3.7683 | 3.9261 | 2.4372 | 1.7875 | | 2.7989 | H11 | 3.1379 | 1.0917 | 2.2013 | 2.0589 | 3.2251 | 3.5853 | 4.0026 | 1.7848 | 2.4306 | 2.7989 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O4 |
C2 |
107.948 |
|
C1 |
O5 |
C3 |
105.253 |
| C2 |
C3 |
O5 |
101.637 |
|
C2 |
C3 |
H9 |
111.086 |
| C2 |
C3 |
H10 |
114.579 |
|
C3 |
C2 |
O4 |
102.856 |
| C3 |
C2 |
H8 |
111.038 |
|
C3 |
C2 |
H11 |
113.767 |
| O4 |
C1 |
O5 |
107.425 |
|
O4 |
C1 |
H6 |
108.636 |
| O4 |
C1 |
H7 |
110.995 |
|
O4 |
C2 |
H8 |
110.287 |
| O4 |
C2 |
H11 |
109.464 |
|
O5 |
C1 |
H6 |
111.158 |
| O5 |
C1 |
H7 |
108.457 |
|
O5 |
C3 |
H9 |
110.786 |
| O5 |
C3 |
H10 |
109.081 |
|
H6 |
C1 |
H7 |
110.147 |
| H8 |
C2 |
H11 |
109.263 |
|
H9 |
C3 |
H10 |
109.436 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.161 |
|
|
|
| 2 |
C |
-0.021 |
|
|
|
| 3 |
C |
-0.108 |
|
|
|
| 4 |
O |
-0.376 |
|
|
|
| 5 |
O |
-0.351 |
|
|
|
| 6 |
H |
0.092 |
|
|
|
| 7 |
H |
0.122 |
|
|
|
| 8 |
H |
0.120 |
|
|
|
| 9 |
H |
0.117 |
|
|
|
| 10 |
H |
0.133 |
|
|
|
| 11 |
H |
0.111 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.232 |
0.366 |
0.836 |
1.533 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.694 |
-0.803 |
0.595 |
| y |
-0.803 |
-34.309 |
0.425 |
| z |
0.595 |
0.425 |
-29.451 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.186 |
-0.803 |
0.595 |
| y |
-0.803 |
-6.736 |
0.425 |
| z |
0.595 |
0.425 |
0.550 |
|
| Polar |
|---|
| 3z2-r2 | 1.100 |
|---|
| x2-y2 | 8.615 |
|---|
| xy | -0.803 |
|---|
| xz | 0.595 |
|---|
| yz | 0.425 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.454 |
-0.088 |
0.068 |
| y |
-0.088 |
5.179 |
0.087 |
| z |
0.068 |
0.087 |
5.132 |
<r2> (average value of r
2) Å
2
| <r2> |
92.717 |
| (<r2>)1/2 |
9.629 |
Jump to
S1C1
S1C2
Energy calculated at wB97X-D/6-311G**
| | hartrees |
|---|
| Energy at 0K | -268.330614 |
| Energy at 298.15K | |
| HF Energy | -268.330614 |
| Nuclear repulsion energy | 195.054913 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at wB97X-D/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3140 |
3004 |
37.27 |
|
|
|
| 2 |
A |
3136 |
2999 |
25.17 |
|
|
|
| 3 |
A |
3058 |
2925 |
77.69 |
|
|
|
| 4 |
A |
3029 |
2897 |
84.93 |
|
|
|
| 5 |
A |
3022 |
2891 |
0.15 |
|
|
|
| 6 |
A |
3014 |
2883 |
141.69 |
|
|
|
| 7 |
A |
1554 |
1486 |
2.22 |
|
|
|
| 8 |
A |
1530 |
1464 |
0.69 |
|
|
|
| 9 |
A |
1524 |
1458 |
3.65 |
|
|
|
| 10 |
A |
1446 |
1383 |
8.63 |
|
|
|
| 11 |
A |
1404 |
1343 |
7.12 |
|
|
|
| 12 |
A |
1359 |
1300 |
0.10 |
|
|
|
| 13 |
A |
1266 |
1211 |
13.20 |
|
|
|
| 14 |
A |
1248 |
1193 |
4.96 |
|
|
|
| 15 |
A |
1232 |
1178 |
83.61 |
|
|
|
| 16 |
A |
1182 |
1130 |
110.19 |
|
|
|
| 17 |
A |
1157 |
1107 |
46.18 |
|
|
|
| 18 |
A |
1155 |
1105 |
21.41 |
|
|
|
| 19 |
A |
1106 |
1058 |
33.80 |
|
|
|
| 20 |
A |
994 |
950 |
3.60 |
|
|
|
| 21 |
A |
985 |
942 |
68.85 |
|
|
|
| 22 |
A |
962 |
920 |
3.60 |
|
|
|
| 23 |
A |
925 |
885 |
22.92 |
|
|
|
| 24 |
A |
746 |
714 |
0.50 |
|
|
|
| 25 |
A |
661 |
633 |
4.38 |
|
|
|
| 26 |
A |
273 |
261 |
0.59 |
|
|
|
| 27 |
A |
117i |
112i |
18.32 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20494.3 cm
-1
Scaled (by 0.9565) Zero Point Vibrational Energy (zpe) 19602.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at wB97X-D/6-311G**
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.201 |
-0.000 |
0.000 |
| C2 |
-0.942 |
0.732 |
0.196 |
| C3 |
-0.942 |
-0.732 |
-0.196 |
| O4 |
0.372 |
1.130 |
-0.172 |
| O5 |
0.371 |
-1.130 |
0.171 |
| H6 |
1.824 |
-0.092 |
-0.896 |
| H7 |
1.824 |
0.092 |
0.897 |
| H8 |
-1.098 |
0.852 |
1.275 |
| H9 |
-1.098 |
-0.852 |
-1.275 |
| H10 |
-1.651 |
-1.350 |
0.353 |
| H11 |
-1.651 |
1.351 |
-0.353 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
O5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 2.2725 | 2.2726 | 1.4123 | 1.4123 | 1.0959 | 1.0959 | 2.7634 | 2.7637 | 3.1753 | 3.1754 |
C2 | 2.2725 | | 1.5151 | 1.4205 | 2.2786 | 3.0858 | 2.9239 | 1.0974 | 2.1669 | 2.2054 | 1.0896 | C3 | 2.2726 | 1.5151 | | 2.2785 | 1.4205 | 2.9242 | 3.0856 | 2.1669 | 1.0973 | 1.0896 | 2.2054 | O4 | 1.4123 | 1.4205 | 2.2785 | | 2.2862 | 2.0322 | 2.0806 | 2.0810 | 2.7028 | 3.2433 | 2.0427 | O5 | 1.4123 | 2.2786 | 1.4205 | 2.2862 | | 2.0806 | 2.0322 | 2.7029 | 2.0810 | 2.0427 | 3.2434 | H6 | 1.0959 | 3.0858 | 2.9242 | 2.0322 | 2.0806 | | 1.8028 | 3.7613 | 3.0434 | 3.9018 | 3.8020 | H7 | 1.0959 | 2.9239 | 3.0856 | 2.0806 | 2.0322 | 1.8028 | | 3.0427 | 3.7614 | 3.8017 | 3.9016 | H8 | 2.7634 | 1.0974 | 2.1669 | 2.0810 | 2.7029 | 3.7613 | 3.0427 | | 3.0669 | 2.4506 | 1.7905 | H9 | 2.7637 | 2.1669 | 1.0973 | 2.7028 | 2.0810 | 3.0434 | 3.7614 | 3.0669 | | 1.7905 | 2.4506 | H10 | 3.1753 | 2.2054 | 1.0896 | 3.2433 | 2.0427 | 3.9018 | 3.8017 | 2.4506 | 1.7905 | | 2.7916 | H11 | 3.1754 | 1.0896 | 2.2054 | 2.0427 | 3.2434 | 3.8020 | 3.9016 | 1.7905 | 2.4506 | 2.7916 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
O4 |
C2 |
106.685 |
|
C1 |
O5 |
C3 |
106.688 |
| C2 |
C3 |
O5 |
101.777 |
|
C2 |
C3 |
H9 |
111.077 |
| C2 |
C3 |
H10 |
114.728 |
|
C3 |
C2 |
O4 |
101.776 |
| C3 |
C2 |
H8 |
111.078 |
|
C3 |
C2 |
H11 |
114.729 |
| O4 |
C1 |
O5 |
108.067 |
|
O4 |
C1 |
H6 |
107.568 |
| O4 |
C1 |
H7 |
111.479 |
|
O4 |
C2 |
H8 |
110.827 |
| O4 |
C2 |
H11 |
108.213 |
|
O5 |
C1 |
H6 |
111.479 |
| O5 |
C1 |
H7 |
107.568 |
|
O5 |
C3 |
H9 |
110.827 |
| O5 |
C3 |
H10 |
108.213 |
|
H6 |
C1 |
H7 |
110.681 |
| H8 |
C2 |
H11 |
109.914 |
|
H9 |
C3 |
H10 |
109.914 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.180 |
|
|
|
| 2 |
C |
-0.056 |
|
|
|
| 3 |
C |
-0.056 |
|
|
|
| 4 |
O |
-0.376 |
|
|
|
| 5 |
O |
-0.376 |
|
|
|
| 6 |
H |
0.104 |
|
|
|
| 7 |
H |
0.104 |
|
|
|
| 8 |
H |
0.114 |
|
|
|
| 9 |
H |
0.114 |
|
|
|
| 10 |
H |
0.125 |
|
|
|
| 11 |
H |
0.125 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.217 |
0.001 |
0.001 |
1.217 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.639 |
-0.001 |
0.000 |
| y |
-0.001 |
-34.481 |
1.050 |
| z |
0.000 |
1.050 |
-29.257 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.230 |
-0.001 |
0.000 |
| y |
-0.001 |
-7.033 |
1.050 |
| z |
0.000 |
1.050 |
0.803 |
|
| Polar |
|---|
| 3z2-r2 | 1.607 |
|---|
| x2-y2 | 8.842 |
|---|
| xy | -0.001 |
|---|
| xz | 0.000 |
|---|
| yz | 1.050 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.522 |
-0.000 |
0.000 |
| y |
-0.000 |
5.143 |
0.107 |
| z |
0.000 |
0.107 |
5.189 |
<r2> (average value of r
2) Å
2
| <r2> |
92.721 |
| (<r2>)1/2 |
9.629 |