Vibrational Frequencies calculated at wB97X-D/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3135 |
2986 |
38.32 |
|
|
|
| 2 |
A' |
3069 |
2922 |
29.58 |
|
|
|
| 3 |
A' |
3050 |
2905 |
26.63 |
|
|
|
| 4 |
A' |
3041 |
2896 |
39.44 |
|
|
|
| 5 |
A' |
3031 |
2886 |
22.87 |
|
|
|
| 6 |
A' |
2928 |
2788 |
140.83 |
|
|
|
| 7 |
A' |
1853 |
1764 |
184.29 |
|
|
|
| 8 |
A' |
1511 |
1439 |
10.46 |
|
|
|
| 9 |
A' |
1498 |
1427 |
1.38 |
|
|
|
| 10 |
A' |
1492 |
1421 |
0.95 |
|
|
|
| 11 |
A' |
1458 |
1389 |
13.29 |
|
|
|
| 12 |
A' |
1432 |
1363 |
14.15 |
|
|
|
| 13 |
A' |
1430 |
1362 |
6.55 |
|
|
|
| 14 |
A' |
1427 |
1359 |
7.51 |
|
|
|
| 15 |
A' |
1384 |
1318 |
10.98 |
|
|
|
| 16 |
A' |
1294 |
1233 |
5.02 |
|
|
|
| 17 |
A' |
1148 |
1093 |
11.29 |
|
|
|
| 18 |
A' |
1090 |
1038 |
0.56 |
|
|
|
| 19 |
A' |
1056 |
1006 |
1.87 |
|
|
|
| 20 |
A' |
931 |
886 |
0.94 |
|
|
|
| 21 |
A' |
908 |
865 |
12.92 |
|
|
|
| 22 |
A' |
697 |
664 |
13.25 |
|
|
|
| 23 |
A' |
404 |
385 |
2.25 |
|
|
|
| 24 |
A' |
298 |
284 |
4.33 |
|
|
|
| 25 |
A' |
141 |
134 |
5.80 |
|
|
|
| 26 |
A" |
3130 |
2981 |
68.33 |
|
|
|
| 27 |
A" |
3117 |
2968 |
16.84 |
|
|
|
| 28 |
A" |
3077 |
2931 |
8.16 |
|
|
|
| 29 |
A" |
3065 |
2919 |
9.74 |
|
|
|
| 30 |
A" |
1513 |
1440 |
9.89 |
|
|
|
| 31 |
A" |
1341 |
1277 |
0.24 |
|
|
|
| 32 |
A" |
1322 |
1259 |
0.14 |
|
|
|
| 33 |
A" |
1244 |
1185 |
0.09 |
|
|
|
| 34 |
A" |
1162 |
1107 |
0.25 |
|
|
|
| 35 |
A" |
988 |
941 |
0.82 |
|
|
|
| 36 |
A" |
864 |
823 |
0.19 |
|
|
|
| 37 |
A" |
744 |
708 |
1.57 |
|
|
|
| 38 |
A" |
661 |
630 |
3.54 |
|
|
|
| 39 |
A" |
246 |
234 |
0.00 |
|
|
|
| 40 |
A" |
160 |
153 |
1.46 |
|
|
|
| 41 |
A" |
110 |
105 |
0.51 |
|
|
|
| 42 |
A" |
53 |
51 |
3.70 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31251.8 cm
-1
Scaled (by 0.9523) Zero Point Vibrational Energy (zpe) 29761.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
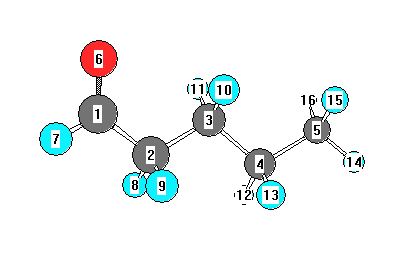
Charges from optimized geometry at wB97X-D/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.074 |
|
|
|
| 2 |
C |
-0.134 |
|
|
|
| 3 |
C |
-0.395 |
|
|
|
| 4 |
C |
-0.137 |
|
|
|
| 5 |
C |
-0.604 |
|
|
|
| 6 |
O |
-0.373 |
|
|
|
| 7 |
H |
0.119 |
|
|
|
| 8 |
H |
0.175 |
|
|
|
| 9 |
H |
0.175 |
|
|
|
| 10 |
H |
0.168 |
|
|
|
| 11 |
H |
0.168 |
|
|
|
| 12 |
H |
0.146 |
|
|
|
| 13 |
H |
0.146 |
|
|
|
| 14 |
H |
0.157 |
|
|
|
| 15 |
H |
0.157 |
|
|
|
| 16 |
H |
0.157 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-2.899 |
-0.151 |
0.000 |
2.903 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.104 |
4.310 |
0.000 |
| y |
4.310 |
-36.688 |
0.000 |
| z |
0.000 |
0.000 |
-37.404 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-9.059 |
4.310 |
0.000 |
| y |
4.310 |
5.067 |
0.000 |
| z |
0.000 |
0.000 |
3.992 |
|
| Polar |
|---|
| 3z2-r2 | 7.983 |
|---|
| x2-y2 | -9.417 |
|---|
| xy | 4.310 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.672 |
-0.390 |
0.000 |
| y |
-0.390 |
10.302 |
0.000 |
| z |
0.000 |
0.000 |
7.416 |
<r2> (average value of r
2) Å
2
| <r2> |
252.201 |
| (<r2>)1/2 |
15.881 |