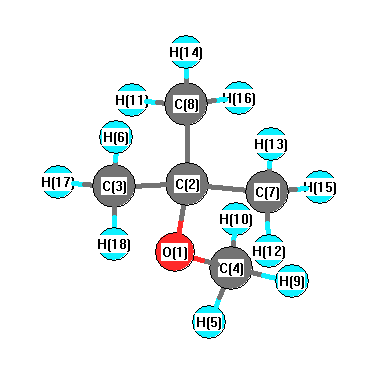
Vibrational Frequencies calculated at wB97X-D/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3159 |
3009 |
42.53 |
|
|
|
| 2 |
A' |
3156 |
3007 |
37.35 |
|
|
|
| 3 |
A' |
3152 |
3002 |
32.48 |
|
|
|
| 4 |
A' |
3147 |
2998 |
34.66 |
|
|
|
| 5 |
A' |
3070 |
2924 |
6.53 |
|
|
|
| 6 |
A' |
3063 |
2918 |
32.63 |
|
|
|
| 7 |
A' |
3021 |
2878 |
60.54 |
|
|
|
| 8 |
A' |
1533 |
1461 |
10.97 |
|
|
|
| 9 |
A' |
1518 |
1446 |
8.27 |
|
|
|
| 10 |
A' |
1504 |
1432 |
5.43 |
|
|
|
| 11 |
A' |
1495 |
1424 |
2.05 |
|
|
|
| 12 |
A' |
1477 |
1407 |
0.30 |
|
|
|
| 13 |
A' |
1435 |
1367 |
16.70 |
|
|
|
| 14 |
A' |
1414 |
1347 |
24.48 |
|
|
|
| 15 |
A' |
1302 |
1240 |
13.99 |
|
|
|
| 16 |
A' |
1258 |
1198 |
124.64 |
|
|
|
| 17 |
A' |
1212 |
1155 |
2.80 |
|
|
|
| 18 |
A' |
1160 |
1105 |
83.40 |
|
|
|
| 19 |
A' |
1052 |
1002 |
1.42 |
|
|
|
| 20 |
A' |
936 |
892 |
0.15 |
|
|
|
| 21 |
A' |
887 |
845 |
19.86 |
|
|
|
| 22 |
A' |
743 |
708 |
4.79 |
|
|
|
| 23 |
A' |
517 |
493 |
1.50 |
|
|
|
| 24 |
A' |
426 |
406 |
0.68 |
|
|
|
| 25 |
A' |
374 |
356 |
0.78 |
|
|
|
| 26 |
A' |
293 |
279 |
0.90 |
|
|
|
| 27 |
A' |
254 |
242 |
0.22 |
|
|
|
| 28 |
A" |
3154 |
3005 |
38.23 |
|
|
|
| 29 |
A" |
3151 |
3001 |
0.28 |
|
|
|
| 30 |
A" |
3143 |
2994 |
0.68 |
|
|
|
| 31 |
A" |
3082 |
2936 |
67.64 |
|
|
|
| 32 |
A" |
3062 |
2917 |
15.62 |
|
|
|
| 33 |
A" |
1517 |
1445 |
0.02 |
|
|
|
| 34 |
A" |
1496 |
1425 |
8.75 |
|
|
|
| 35 |
A" |
1489 |
1418 |
0.35 |
|
|
|
| 36 |
A" |
1477 |
1407 |
0.01 |
|
|
|
| 37 |
A" |
1407 |
1340 |
25.57 |
|
|
|
| 38 |
A" |
1270 |
1209 |
19.26 |
|
|
|
| 39 |
A" |
1168 |
1112 |
2.14 |
|
|
|
| 40 |
A" |
1041 |
992 |
2.15 |
|
|
|
| 41 |
A" |
960 |
914 |
0.03 |
|
|
|
| 42 |
A" |
923 |
879 |
0.02 |
|
|
|
| 43 |
A" |
469 |
447 |
6.35 |
|
|
|
| 44 |
A" |
350 |
333 |
1.31 |
|
|
|
| 45 |
A" |
277 |
264 |
1.21 |
|
|
|
| 46 |
A" |
214 |
204 |
1.70 |
|
|
|
| 47 |
A" |
142 |
135 |
3.11 |
|
|
|
| 48 |
A" |
104i |
99i |
2.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 36122.0 cm
-1
Scaled (by 0.9526) Zero Point Vibrational Energy (zpe) 34409.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.275 |
|
|
|
| 2 |
C |
-0.101 |
|
|
|
| 3 |
C |
-0.449 |
|
|
|
| 4 |
C |
-0.178 |
|
|
|
| 5 |
H |
0.156 |
|
|
|
| 6 |
H |
0.150 |
|
|
|
| 7 |
C |
-0.432 |
|
|
|
| 8 |
C |
-0.432 |
|
|
|
| 9 |
H |
0.130 |
|
|
|
| 10 |
H |
0.130 |
|
|
|
| 11 |
H |
0.170 |
|
|
|
| 12 |
H |
0.170 |
|
|
|
| 13 |
H |
0.160 |
|
|
|
| 14 |
H |
0.160 |
|
|
|
| 15 |
H |
0.152 |
|
|
|
| 16 |
H |
0.152 |
|
|
|
| 17 |
H |
0.168 |
|
|
|
| 18 |
H |
0.168 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.255 |
0.561 |
0.000 |
1.374 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.643 |
-2.157 |
0.000 |
| y |
-2.157 |
-38.365 |
0.000 |
| z |
0.000 |
0.000 |
-40.039 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.441 |
-2.157 |
0.000 |
| y |
-2.157 |
1.976 |
0.000 |
| z |
0.000 |
0.000 |
-0.535 |
|
| Polar |
|---|
| 3z2-r2 | -1.069 |
|---|
| x2-y2 | -2.278 |
|---|
| xy | -2.157 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.266 |
-0.405 |
0.000 |
| y |
-0.405 |
10.003 |
0.000 |
| z |
0.000 |
0.000 |
9.271 |
<r2> (average value of r
2) Å
2
| <r2> |
179.780 |
| (<r2>)1/2 |
13.408 |