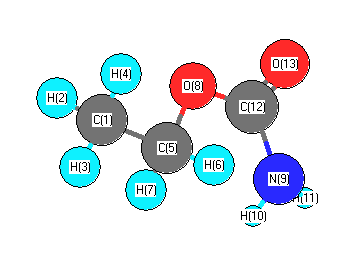
Vibrational Frequencies calculated at wB97X-D/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3782 |
3603 |
55.35 |
|
|
|
| 2 |
A |
3654 |
3481 |
35.75 |
|
|
|
| 3 |
A |
3175 |
3025 |
19.28 |
|
|
|
| 4 |
A |
3164 |
3014 |
19.05 |
|
|
|
| 5 |
A |
3095 |
2948 |
23.92 |
|
|
|
| 6 |
A |
3078 |
2932 |
15.74 |
|
|
|
| 7 |
A |
3030 |
2887 |
41.44 |
|
|
|
| 8 |
A |
1865 |
1777 |
632.76 |
|
|
|
| 9 |
A |
1659 |
1580 |
153.29 |
|
|
|
| 10 |
A |
1526 |
1454 |
1.37 |
|
|
|
| 11 |
A |
1507 |
1435 |
4.09 |
|
|
|
| 12 |
A |
1495 |
1424 |
8.60 |
|
|
|
| 13 |
A |
1452 |
1383 |
12.21 |
|
|
|
| 14 |
A |
1419 |
1352 |
81.01 |
|
|
|
| 15 |
A |
1372 |
1307 |
372.19 |
|
|
|
| 16 |
A |
1321 |
1258 |
1.26 |
|
|
|
| 17 |
A |
1182 |
1126 |
5.48 |
|
|
|
| 18 |
A |
1175 |
1119 |
56.69 |
|
|
|
| 19 |
A |
1136 |
1082 |
94.49 |
|
|
|
| 20 |
A |
1112 |
1059 |
15.44 |
|
|
|
| 21 |
A |
1010 |
962 |
9.49 |
|
|
|
| 22 |
A |
880 |
838 |
10.14 |
|
|
|
| 23 |
A |
845 |
805 |
0.41 |
|
|
|
| 24 |
A |
777 |
740 |
29.07 |
|
|
|
| 25 |
A |
584 |
556 |
9.35 |
|
|
|
| 26 |
A |
568 |
541 |
7.22 |
|
|
|
| 27 |
A |
529 |
504 |
62.76 |
|
|
|
| 28 |
A |
398 |
379 |
14.80 |
|
|
|
| 29 |
A |
352 |
335 |
163.07 |
|
|
|
| 30 |
A |
248 |
237 |
0.46 |
|
|
|
| 31 |
A |
223 |
213 |
2.87 |
|
|
|
| 32 |
A |
92 |
88 |
1.88 |
|
|
|
| 33 |
A |
79 |
75 |
0.20 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23891.7 cm
-1
Scaled (by 0.9526) Zero Point Vibrational Energy (zpe) 22759.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at wB97X-D/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.625 |
|
|
|
| 2 |
H |
0.181 |
|
|
|
| 3 |
H |
0.162 |
|
|
|
| 4 |
H |
0.183 |
|
|
|
| 5 |
C |
-0.006 |
|
|
|
| 6 |
H |
0.163 |
|
|
|
| 7 |
H |
0.137 |
|
|
|
| 8 |
O |
-0.381 |
|
|
|
| 9 |
N |
-0.638 |
|
|
|
| 10 |
H |
0.299 |
|
|
|
| 11 |
H |
0.345 |
|
|
|
| 12 |
C |
0.696 |
|
|
|
| 13 |
O |
-0.515 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.704 |
4.688 |
-0.543 |
5.439 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.054 |
-6.329 |
0.944 |
| y |
-6.329 |
-36.490 |
-1.372 |
| z |
0.944 |
-1.372 |
-36.624 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.503 |
-6.329 |
0.944 |
| y |
-6.329 |
-0.151 |
-1.372 |
| z |
0.944 |
-1.372 |
-0.352 |
|
| Polar |
|---|
| 3z2-r2 | -0.704 |
|---|
| x2-y2 | 0.435 |
|---|
| xy | -6.329 |
|---|
| xz | 0.944 |
|---|
| yz | -1.372 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.098 |
0.382 |
-0.075 |
| y |
0.382 |
7.719 |
-0.016 |
| z |
-0.075 |
-0.016 |
5.811 |
<r2> (average value of r
2) Å
2
| <r2> |
192.108 |
| (<r2>)1/2 |
13.860 |