Jump to
S1C2
S1C3
Energy calculated at PBEPBE/3-21G
| | hartrees |
|---|
| Energy at 0K | -577.925230 |
| Energy at 298.15K | |
| HF Energy | -577.925230 |
| Nuclear repulsion energy | 77.831476 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2175 |
2156 |
0.00 |
346.94 |
0.17 |
0.29 |
| 2 |
Ag |
883 |
875 |
0.00 |
30.02 |
0.70 |
0.82 |
| 3 |
Ag |
567 |
562 |
0.00 |
58.97 |
0.28 |
0.44 |
| 4 |
Au |
540 |
535 |
0.00 |
0.00 |
0.00 |
0.00 |
| 5 |
B1u |
2169 |
2149 |
59.30 |
0.00 |
0.00 |
0.00 |
| 6 |
B1u |
808 |
801 |
95.23 |
0.00 |
0.00 |
0.00 |
| 7 |
B2g |
193i |
192i |
0.00 |
51.14 |
0.75 |
0.86 |
| 8 |
B2u |
2215 |
2195 |
130.12 |
0.00 |
0.00 |
0.00 |
| 9 |
B2u |
354 |
351 |
10.56 |
0.00 |
0.00 |
0.00 |
| 10 |
B3g |
2202 |
2182 |
0.00 |
172.50 |
0.75 |
0.86 |
| 11 |
B3g |
563 |
558 |
0.00 |
7.24 |
0.75 |
0.86 |
| 12 |
B3u |
478 |
474 |
0.00 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 6380.4 cm
-1
Scaled (by 0.9909) Zero Point Vibrational Energy (zpe) 6322.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/3-21G
Point Group is D2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
0.000 |
1.082 |
| Si2 |
0.000 |
0.000 |
-1.082 |
| H3 |
0.000 |
1.275 |
1.873 |
| H4 |
0.000 |
-1.275 |
1.873 |
| H5 |
0.000 |
1.275 |
-1.873 |
| H6 |
0.000 |
-1.275 |
-1.873 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1633 | 1.5006 | 1.5006 | 3.2180 | 3.2180 |
Si2 | 2.1633 | | 3.2180 | 3.2180 | 1.5006 | 1.5006 | H3 | 1.5006 | 3.2180 | | 2.5501 | 3.7460 | 4.5316 | H4 | 1.5006 | 3.2180 | 2.5501 | | 4.5316 | 3.7460 | H5 | 3.2180 | 1.5006 | 3.7460 | 4.5316 | | 2.5501 | H6 | 3.2180 | 1.5006 | 4.5316 | 3.7460 | 2.5501 | |
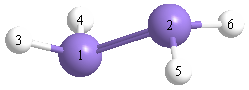 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
121.826 |
|
Si1 |
Si2 |
H6 |
121.826 |
| Si2 |
Si1 |
H3 |
121.826 |
|
Si2 |
Si1 |
H4 |
121.826 |
| H3 |
Si1 |
H4 |
116.348 |
|
H5 |
Si2 |
H6 |
116.348 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.083 |
|
|
|
| 2 |
Si |
0.083 |
|
|
|
| 3 |
H |
-0.042 |
|
|
|
| 4 |
H |
-0.042 |
|
|
|
| 5 |
H |
-0.042 |
|
|
|
| 6 |
H |
-0.042 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.209 |
0.000 |
0.000 |
| y |
0.000 |
-29.621 |
0.000 |
| z |
0.000 |
0.000 |
-28.097 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.350 |
0.000 |
0.000 |
| y |
0.000 |
0.532 |
0.000 |
| z |
0.000 |
0.000 |
2.818 |
|
| Polar |
|---|
| 3z2-r2 | 5.636 |
|---|
| x2-y2 | -2.588 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.338 |
0.000 |
0.000 |
| y |
0.000 |
6.101 |
0.000 |
| z |
0.000 |
0.000 |
13.600 |
<r2> (average value of r
2) Å
2
| <r2> |
72.017 |
| (<r2>)1/2 |
8.486 |
Jump to
S1C1
S1C3
Energy calculated at PBEPBE/3-21G
| | hartrees |
|---|
| Energy at 0K | -577.925895 |
| Energy at 298.15K | |
| HF Energy | -577.925895 |
| Nuclear repulsion energy | 77.322860 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
2150 |
2130 |
0.00 |
349.59 |
0.15 |
0.26 |
| 2 |
Ag |
886 |
878 |
0.00 |
41.86 |
0.66 |
0.80 |
| 3 |
Ag |
548 |
543 |
0.00 |
51.10 |
0.37 |
0.54 |
| 4 |
Ag |
265 |
263 |
0.00 |
37.18 |
0.35 |
0.51 |
| 5 |
Au |
2189 |
2169 |
152.98 |
0.00 |
0.00 |
0.00 |
| 6 |
Au |
523 |
518 |
0.03 |
0.00 |
0.00 |
0.00 |
| 7 |
Au |
342 |
339 |
10.66 |
0.00 |
0.00 |
0.00 |
| 8 |
Bg |
2175 |
2155 |
0.00 |
204.47 |
0.75 |
0.86 |
| 9 |
Bg |
578 |
573 |
0.00 |
12.04 |
0.75 |
0.86 |
| 10 |
Bu |
2145 |
2126 |
91.21 |
0.00 |
0.00 |
0.00 |
| 11 |
Bu |
848 |
840 |
139.85 |
0.00 |
0.00 |
0.00 |
| 12 |
Bu |
427 |
423 |
17.82 |
0.00 |
0.00 |
0.00 |
Unscaled Zero Point Vibrational Energy (zpe) 6537.9 cm
-1
Scaled (by 0.9909) Zero Point Vibrational Energy (zpe) 6478.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/3-21G
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.093 |
0.000 |
| Si2 |
0.000 |
-1.093 |
0.000 |
| H3 |
0.381 |
1.828 |
1.258 |
| H4 |
0.381 |
1.828 |
-1.258 |
| H5 |
-0.381 |
-1.828 |
1.258 |
| H6 |
-0.381 |
-1.828 |
-1.258 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.1856 | 1.5060 | 1.5060 | 3.2032 | 3.2032 |
Si2 | 2.1856 | | 3.2032 | 3.2032 | 1.5060 | 1.5060 | H3 | 1.5060 | 3.2032 | | 2.5155 | 3.7353 | 4.5033 | H4 | 1.5060 | 3.2032 | 2.5155 | | 4.5033 | 3.7353 | H5 | 3.2032 | 1.5060 | 3.7353 | 4.5033 | | 2.5155 | H6 | 3.2032 | 1.5060 | 4.5033 | 3.7353 | 2.5155 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
119.241 |
|
Si1 |
Si2 |
H6 |
119.241 |
| Si2 |
Si1 |
H3 |
119.241 |
|
Si2 |
Si1 |
H4 |
119.241 |
| H3 |
Si1 |
H4 |
113.266 |
|
H5 |
Si2 |
H6 |
113.266 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.090 |
|
|
|
| 2 |
Si |
0.090 |
|
|
|
| 3 |
H |
-0.045 |
|
|
|
| 4 |
H |
-0.045 |
|
|
|
| 5 |
H |
-0.045 |
|
|
|
| 6 |
H |
-0.045 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.020 |
0.178 |
0.000 |
| y |
0.178 |
-28.307 |
0.000 |
| z |
0.000 |
0.000 |
-29.877 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.928 |
0.178 |
0.000 |
| y |
0.178 |
2.641 |
0.000 |
| z |
0.000 |
0.000 |
0.286 |
|
| Polar |
|---|
| 3z2-r2 | 0.572 |
|---|
| x2-y2 | -3.713 |
|---|
| xy | 0.178 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.537 |
-0.629 |
0.000 |
| y |
-0.629 |
13.709 |
0.000 |
| z |
0.000 |
0.000 |
6.470 |
<r2> (average value of r
2) Å
2
| <r2> |
72.499 |
| (<r2>)1/2 |
8.515 |
Jump to
S1C1
S1C2
Energy calculated at PBEPBE/3-21G
| | hartrees |
|---|
| Energy at 0K | -577.872479 |
| Energy at 298.15K | |
| HF Energy | -577.872479 |
| Nuclear repulsion energy | 69.182443 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
1932 |
1914 |
388.09 |
248.96 |
0.41 |
0.58 |
| 2 |
A1 |
1404 |
1391 |
0.89 |
57.39 |
0.16 |
0.27 |
| 3 |
A1 |
864 |
856 |
33.82 |
101.04 |
0.42 |
0.59 |
| 4 |
A1 |
442 |
438 |
0.32 |
33.50 |
0.44 |
0.61 |
| 5 |
A1 |
345 |
342 |
0.60 |
25.72 |
0.28 |
0.43 |
| 6 |
A2 |
1092 |
1082 |
0.00 |
4.52 |
0.75 |
0.86 |
| 7 |
A2 |
598 |
592 |
0.00 |
0.12 |
0.75 |
0.86 |
| 8 |
B1 |
1053 |
1044 |
59.91 |
2.07 |
0.75 |
0.86 |
| 9 |
B1 |
784 |
777 |
19.48 |
32.31 |
0.75 |
0.86 |
| 10 |
B2 |
1906 |
1889 |
5.42 |
4.35 |
0.75 |
0.86 |
| 11 |
B2 |
1039 |
1030 |
848.86 |
13.82 |
0.75 |
0.86 |
| 12 |
B2 |
681 |
675 |
19.35 |
25.89 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 6070.1 cm
-1
Scaled (by 0.9909) Zero Point Vibrational Energy (zpe) 6014.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/3-21G
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Si1 |
0.000 |
1.407 |
-0.089 |
| Si2 |
0.000 |
-1.407 |
-0.089 |
| H3 |
-1.047 |
0.000 |
-0.231 |
| H4 |
1.047 |
0.000 |
-0.231 |
| H5 |
0.000 |
1.351 |
1.473 |
| H6 |
0.000 |
-1.351 |
1.473 |
Atom - Atom Distances (Å)
| |
Si1 |
Si2 |
H3 |
H4 |
H5 |
H6 |
| Si1 | | 2.8140 | 1.7598 | 1.7598 | 1.5624 | 3.1691 |
Si2 | 2.8140 | | 1.7598 | 1.7598 | 3.1691 | 1.5624 | H3 | 1.7598 | 1.7598 | | 2.0946 | 2.4133 | 2.4133 | H4 | 1.7598 | 1.7598 | 2.0946 | | 2.4133 | 2.4133 | H5 | 1.5624 | 3.1691 | 2.4133 | 2.4133 | | 2.7014 | H6 | 3.1691 | 1.5624 | 2.4133 | 2.4133 | 2.7014 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Si1 |
Si2 |
H5 |
29.518 |
|
Si1 |
Si2 |
H6 |
87.936 |
| Si2 |
Si1 |
H3 |
36.914 |
|
Si2 |
Si1 |
H4 |
36.914 |
| H3 |
Si1 |
H4 |
73.046 |
|
H5 |
Si2 |
H6 |
58.418 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Si |
0.232 |
|
|
|
| 2 |
Si |
0.232 |
|
|
|
| 3 |
H |
-0.163 |
|
|
|
| 4 |
H |
-0.163 |
|
|
|
| 5 |
H |
-0.069 |
|
|
|
| 6 |
H |
-0.069 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.985 |
0.985 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.069 |
0.000 |
0.000 |
| y |
0.000 |
-32.599 |
0.000 |
| z |
0.000 |
0.000 |
-34.348 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.405 |
0.000 |
0.000 |
| y |
0.000 |
-0.891 |
0.000 |
| z |
0.000 |
0.000 |
-3.514 |
|
| Polar |
|---|
| 3z2-r2 | -7.029 |
|---|
| x2-y2 | 3.530 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.854 |
0.000 |
0.000 |
| y |
0.000 |
14.676 |
0.000 |
| z |
0.000 |
0.000 |
7.290 |
<r2> (average value of r
2) Å
2
| <r2> |
85.927 |
| (<r2>)1/2 |
9.270 |