Jump to
S1C2
Vibrational Frequencies calculated at PBEPBE/3-21G
Geometric Data calculated at PBEPBE/3-21G
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at PBEPBE/3-21G
| | hartrees |
|---|
| Energy at 0K | -621.319271 |
| Energy at 298.15K | -621.322581 |
| HF Energy | -621.319271 |
| Nuclear repulsion energy | 176.922355 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3213 |
3183 |
3.80 |
|
|
|
| 2 |
A' |
1143 |
1133 |
19.28 |
|
|
|
| 3 |
A' |
959 |
951 |
29.48 |
|
|
|
| 4 |
A' |
604 |
598 |
56.72 |
|
|
|
| 5 |
A' |
457 |
453 |
130.12 |
|
|
|
| 6 |
A' |
304 |
301 |
65.50 |
|
|
|
| 7 |
A' |
205 |
203 |
7.44 |
|
|
|
| 8 |
A" |
3210 |
3181 |
11.13 |
|
|
|
| 9 |
A" |
1083 |
1073 |
59.98 |
|
|
|
| 10 |
A" |
638 |
632 |
185.61 |
|
|
|
| 11 |
A" |
420 |
417 |
68.43 |
|
|
|
| 12 |
A" |
180 |
178 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 6208.4 cm
-1
Scaled (by 0.9909) Zero Point Vibrational Energy (zpe) 6151.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/3-21G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| S1 |
0.334 |
0.378 |
0.000 |
| O2 |
-1.172 |
0.932 |
0.000 |
| O3 |
0.334 |
-0.729 |
1.419 |
| O4 |
0.334 |
-0.729 |
-1.419 |
| H5 |
-0.660 |
-0.916 |
1.577 |
| H6 |
-0.660 |
-0.916 |
-1.577 |
Atom - Atom Distances (Å)
| |
S1 |
O2 |
O3 |
O4 |
H5 |
H6 |
| S1 | | 1.6047 | 1.8000 | 1.8000 | 2.2687 | 2.2687 |
O2 | 1.6047 | | 2.6535 | 2.6535 | 2.4820 | 2.4820 | O3 | 1.8000 | 2.6535 | | 2.8388 | 1.0236 | 3.1621 | O4 | 1.8000 | 2.6535 | 2.8388 | | 3.1621 | 1.0236 | H5 | 2.2687 | 2.4820 | 1.0236 | 3.1621 | | 3.1533 | H6 | 2.2687 | 2.4820 | 3.1621 | 1.0236 | 3.1533 | |
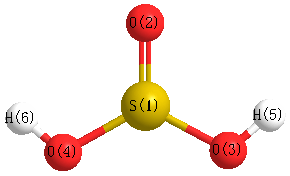 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| S1 |
O3 |
H5 |
103.483 |
|
S1 |
O4 |
H6 |
103.483 |
| O2 |
S1 |
O3 |
102.256 |
|
O2 |
S1 |
O4 |
102.256 |
| O3 |
S1 |
O4 |
104.103 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
S |
0.986 |
|
|
|
| 2 |
O |
-0.562 |
|
|
|
| 3 |
O |
-0.543 |
|
|
|
| 4 |
O |
-0.543 |
|
|
|
| 5 |
H |
0.331 |
|
|
|
| 6 |
H |
0.331 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.105 |
-0.723 |
0.000 |
1.321 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.503 |
5.555 |
0.000 |
| y |
5.555 |
-30.043 |
0.000 |
| z |
0.000 |
0.000 |
-29.791 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.586 |
5.555 |
0.000 |
| y |
5.555 |
0.104 |
0.000 |
| z |
0.000 |
0.000 |
0.482 |
|
| Polar |
|---|
| 3z2-r2 | 0.964 |
|---|
| x2-y2 | -0.461 |
|---|
| xy | 5.555 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.891 |
-0.458 |
0.000 |
| y |
-0.458 |
3.541 |
0.000 |
| z |
0.000 |
0.000 |
4.866 |
<r2> (average value of r
2) Å
2
| <r2> |
90.855 |
| (<r2>)1/2 |
9.532 |