Jump to
S1C2
Energy calculated at PBEPBE/STO-3G
| | hartrees |
|---|
| Energy at 0K | -571.944601 |
| Energy at 298.15K | -571.951955 |
| Nuclear repulsion energy | 155.017127 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3422 |
3126 |
0.75 |
|
|
|
| 2 |
A' |
3291 |
3007 |
1.41 |
|
|
|
| 3 |
A' |
3272 |
2989 |
1.16 |
|
|
|
| 4 |
A' |
3253 |
2972 |
1.61 |
|
|
|
| 5 |
A' |
1651 |
1508 |
4.10 |
|
|
|
| 6 |
A' |
1622 |
1482 |
1.37 |
|
|
|
| 7 |
A' |
1589 |
1452 |
0.83 |
|
|
|
| 8 |
A' |
1530 |
1398 |
0.46 |
|
|
|
| 9 |
A' |
1451 |
1326 |
2.87 |
|
|
|
| 10 |
A' |
1335 |
1220 |
20.94 |
|
|
|
| 11 |
A' |
1173 |
1072 |
0.58 |
|
|
|
| 12 |
A' |
1102 |
1007 |
1.59 |
|
|
|
| 13 |
A' |
966 |
882 |
15.26 |
|
|
|
| 14 |
A' |
823 |
752 |
19.98 |
|
|
|
| 15 |
A' |
365 |
333 |
1.80 |
|
|
|
| 16 |
A' |
228 |
209 |
2.45 |
|
|
|
| 17 |
A" |
3422 |
3127 |
2.06 |
|
|
|
| 18 |
A" |
3401 |
3107 |
0.31 |
|
|
|
| 19 |
A" |
3386 |
3093 |
0.05 |
|
|
|
| 20 |
A" |
1643 |
1501 |
5.55 |
|
|
|
| 21 |
A" |
1378 |
1259 |
0.05 |
|
|
|
| 22 |
A" |
1309 |
1196 |
0.25 |
|
|
|
| 23 |
A" |
1133 |
1035 |
0.00 |
|
|
|
| 24 |
A" |
912 |
833 |
1.28 |
|
|
|
| 25 |
A" |
780 |
712 |
6.59 |
|
|
|
| 26 |
A" |
214 |
195 |
0.06 |
|
|
|
| 27 |
A" |
106 |
96 |
1.71 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22378.1 cm
-1
Scaled (by 0.9136) Zero Point Vibrational Energy (zpe) 20444.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/STO-3G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.603 |
0.000 |
| C2 |
0.934 |
-0.640 |
0.000 |
| C3 |
2.430 |
-0.206 |
0.000 |
| Cl4 |
-1.785 |
0.108 |
0.000 |
| H5 |
0.156 |
1.227 |
0.906 |
| H6 |
0.156 |
1.227 |
-0.906 |
| H7 |
0.719 |
-1.257 |
-0.896 |
| H8 |
0.719 |
-1.257 |
0.896 |
| H9 |
3.083 |
-1.099 |
0.000 |
| H10 |
2.664 |
0.397 |
-0.897 |
| H11 |
2.664 |
0.397 |
0.897 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5551 | 2.5608 | 1.8523 | 1.1115 | 1.1115 | 2.1860 | 2.1860 | 3.5209 | 2.8184 | 2.8184 |
C2 | 1.5551 | | 1.5570 | 2.8204 | 2.2170 | 2.2170 | 1.1091 | 1.1091 | 2.1964 | 2.2071 | 2.2071 | C3 | 2.5608 | 1.5570 | | 4.2263 | 2.8365 | 2.8365 | 2.1991 | 2.1991 | 1.1059 | 1.1061 | 1.1061 | Cl4 | 1.8523 | 2.8204 | 4.2263 | | 2.4169 | 2.4169 | 2.9888 | 2.9888 | 5.0147 | 4.5476 | 4.5476 | H5 | 1.1115 | 2.2170 | 2.8365 | 2.4169 | | 1.8122 | 3.1205 | 2.5476 | 3.8466 | 3.1988 | 2.6420 | H6 | 1.1115 | 2.2170 | 2.8365 | 2.4169 | 1.8122 | | 2.5476 | 3.1205 | 3.8466 | 2.6420 | 3.1988 | H7 | 2.1860 | 1.1091 | 2.1991 | 2.9888 | 3.1205 | 2.5476 | | 1.7918 | 2.5331 | 2.5535 | 3.1203 | H8 | 2.1860 | 1.1091 | 2.1991 | 2.9888 | 2.5476 | 3.1205 | 1.7918 | | 2.5331 | 3.1203 | 2.5535 | H9 | 3.5209 | 2.1964 | 1.1059 | 5.0147 | 3.8466 | 3.8466 | 2.5331 | 2.5331 | | 1.7935 | 1.7935 | H10 | 2.8184 | 2.2071 | 1.1061 | 4.5476 | 3.1988 | 2.6420 | 2.5535 | 3.1203 | 1.7935 | | 1.7946 | H11 | 2.8184 | 2.2071 | 1.1061 | 4.5476 | 2.6420 | 3.1988 | 3.1203 | 2.5535 | 1.7935 | 1.7946 | |
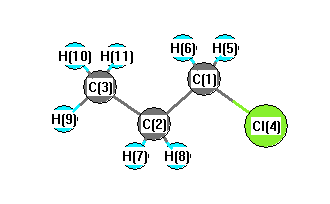 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
110.741 |
|
C1 |
C2 |
H7 |
109.129 |
| C1 |
C2 |
H8 |
109.129 |
|
C2 |
C1 |
Cl4 |
111.433 |
| C2 |
C1 |
H5 |
111.402 |
|
C2 |
C1 |
H6 |
111.402 |
| C2 |
C3 |
H9 |
109.991 |
|
C2 |
C3 |
H10 |
110.814 |
| C2 |
C3 |
H11 |
110.814 |
|
C3 |
C2 |
H7 |
110.008 |
| C3 |
C2 |
H8 |
110.008 |
|
Cl4 |
C1 |
H5 |
106.577 |
| Cl4 |
C1 |
H6 |
106.577 |
|
H5 |
C1 |
H6 |
109.215 |
| H7 |
C2 |
H8 |
107.765 |
|
H9 |
C3 |
H10 |
108.357 |
| H9 |
C3 |
H11 |
108.357 |
|
H10 |
C3 |
H11 |
108.431 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.107 |
|
|
|
| 2 |
C |
-0.138 |
|
|
|
| 3 |
C |
-0.233 |
|
|
|
| 4 |
Cl |
-0.167 |
|
|
|
| 5 |
H |
0.107 |
|
|
|
| 6 |
H |
0.107 |
|
|
|
| 7 |
H |
0.091 |
|
|
|
| 8 |
H |
0.091 |
|
|
|
| 9 |
H |
0.087 |
|
|
|
| 10 |
H |
0.082 |
|
|
|
| 11 |
H |
0.082 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.434 |
0.385 |
0.000 |
2.464 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-33.866 |
0.091 |
0.000 |
| y |
0.091 |
-29.914 |
0.000 |
| z |
0.000 |
0.000 |
-30.204 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.806 |
0.091 |
0.000 |
| y |
0.091 |
2.120 |
0.000 |
| z |
0.000 |
0.000 |
1.686 |
|
| Polar |
|---|
| 3z2-r2 | 3.372 |
|---|
| x2-y2 | -3.951 |
|---|
| xy | 0.091 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.532 |
0.185 |
0.000 |
| y |
0.185 |
2.495 |
0.000 |
| z |
0.000 |
0.000 |
2.379 |
<r2> (average value of r
2) Å
2
| <r2> |
156.810 |
| (<r2>)1/2 |
12.522 |
Jump to
S1C1
Energy calculated at PBEPBE/STO-3G
| | hartrees |
|---|
| Energy at 0K | -571.944622 |
| Energy at 298.15K | -571.952101 |
| Nuclear repulsion energy | 159.370804 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3425 |
3129 |
0.16 |
|
|
|
| 2 |
A |
3422 |
3126 |
1.63 |
|
|
|
| 3 |
A |
3394 |
3101 |
0.45 |
|
|
|
| 4 |
A |
3375 |
3084 |
0.26 |
|
|
|
| 5 |
A |
3273 |
2990 |
1.54 |
|
|
|
| 6 |
A |
3270 |
2987 |
1.55 |
|
|
|
| 7 |
A |
3254 |
2973 |
1.56 |
|
|
|
| 8 |
A |
1650 |
1508 |
4.70 |
|
|
|
| 9 |
A |
1640 |
1498 |
5.09 |
|
|
|
| 10 |
A |
1607 |
1468 |
0.65 |
|
|
|
| 11 |
A |
1583 |
1446 |
1.75 |
|
|
|
| 12 |
A |
1531 |
1398 |
2.12 |
|
|
|
| 13 |
A |
1460 |
1334 |
1.61 |
|
|
|
| 14 |
A |
1396 |
1275 |
21.66 |
|
|
|
| 15 |
A |
1341 |
1225 |
4.38 |
|
|
|
| 16 |
A |
1289 |
1178 |
1.15 |
|
|
|
| 17 |
A |
1163 |
1063 |
1.63 |
|
|
|
| 18 |
A |
1129 |
1031 |
1.60 |
|
|
|
| 19 |
A |
1107 |
1011 |
2.13 |
|
|
|
| 20 |
A |
957 |
874 |
10.83 |
|
|
|
| 21 |
A |
909 |
831 |
6.42 |
|
|
|
| 22 |
A |
841 |
769 |
17.36 |
|
|
|
| 23 |
A |
732 |
669 |
5.03 |
|
|
|
| 24 |
A |
428 |
391 |
1.25 |
|
|
|
| 25 |
A |
283 |
259 |
1.04 |
|
|
|
| 26 |
A |
202 |
185 |
1.22 |
|
|
|
| 27 |
A |
125 |
114 |
1.33 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22392.5 cm
-1
Scaled (by 0.9136) Zero Point Vibrational Energy (zpe) 20457.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/STO-3G
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.174 |
0.916 |
0.313 |
| C2 |
-1.184 |
0.574 |
-0.364 |
| C3 |
-1.805 |
-0.752 |
0.151 |
| Cl4 |
1.472 |
-0.351 |
-0.070 |
| H5 |
0.572 |
1.888 |
-0.049 |
| H6 |
0.081 |
0.944 |
1.420 |
| H7 |
-1.872 |
1.423 |
-0.155 |
| H8 |
-1.038 |
0.526 |
-1.463 |
| H9 |
-2.759 |
-0.956 |
-0.370 |
| H10 |
-1.113 |
-1.594 |
-0.035 |
| H11 |
-2.004 |
-0.696 |
1.238 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
Cl4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
H11 |
| C1 | | 1.5554 | 2.5934 | 1.8536 | 1.1116 | 1.1116 | 2.1593 | 2.1848 | 3.5456 | 2.8416 | 2.8628 |
C2 | 1.5554 | | 1.5529 | 2.8286 | 2.2161 | 2.2186 | 1.1119 | 1.1094 | 2.1960 | 2.1944 | 2.2025 | C3 | 2.5934 | 1.5529 | | 3.3097 | 3.5590 | 2.8368 | 2.1976 | 2.1973 | 1.1057 | 1.1059 | 1.1060 | Cl4 | 1.8536 | 2.8286 | 3.3097 | | 2.4133 | 2.4146 | 3.7866 | 3.0021 | 4.2850 | 2.8687 | 3.7302 | H5 | 1.1116 | 2.2161 | 3.5590 | 2.4133 | | 1.8142 | 2.4906 | 2.5393 | 4.3922 | 3.8685 | 3.8691 | H6 | 1.1116 | 2.2186 | 2.8368 | 2.4146 | 1.8142 | | 2.5549 | 3.1208 | 3.8578 | 3.1598 | 2.6590 | H7 | 2.1593 | 1.1119 | 2.1976 | 3.7866 | 2.4906 | 2.5549 | | 1.7913 | 2.5479 | 3.1132 | 2.5388 | H8 | 2.1848 | 1.1094 | 2.1973 | 3.0021 | 2.5393 | 3.1208 | 1.7913 | | 2.5204 | 2.5571 | 3.1175 | H9 | 3.5456 | 2.1960 | 1.1057 | 4.2850 | 4.3922 | 3.8578 | 2.5479 | 2.5204 | | 1.7970 | 1.7952 | H10 | 2.8416 | 2.1944 | 1.1059 | 2.8687 | 3.8685 | 3.1598 | 3.1132 | 2.5571 | 1.7970 | | 1.7950 | H11 | 2.8628 | 2.2025 | 1.1060 | 3.7302 | 3.8691 | 2.6590 | 2.5388 | 3.1175 | 1.7952 | 1.7950 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
113.095 |
|
C1 |
C2 |
H7 |
106.926 |
| C1 |
C2 |
H8 |
108.992 |
|
C2 |
C1 |
Cl4 |
111.846 |
| C2 |
C1 |
H5 |
111.304 |
|
C2 |
C1 |
H6 |
111.493 |
| C2 |
C3 |
H9 |
110.243 |
|
C2 |
C3 |
H10 |
110.110 |
| C2 |
C3 |
H11 |
110.734 |
|
C3 |
C2 |
H7 |
110.007 |
| C3 |
C2 |
H8 |
110.126 |
|
Cl4 |
C1 |
H5 |
106.238 |
| Cl4 |
C1 |
H6 |
106.326 |
|
H5 |
C1 |
H6 |
109.377 |
| H7 |
C2 |
H8 |
107.499 |
|
H9 |
C3 |
H10 |
108.693 |
| H9 |
C3 |
H11 |
108.519 |
|
H10 |
C3 |
H11 |
108.486 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.108 |
|
|
|
| 2 |
C |
-0.136 |
|
|
|
| 3 |
C |
-0.234 |
|
|
|
| 4 |
Cl |
-0.169 |
|
|
|
| 5 |
H |
0.108 |
|
|
|
| 6 |
H |
0.107 |
|
|
|
| 7 |
H |
0.087 |
|
|
|
| 8 |
H |
0.091 |
|
|
|
| 9 |
H |
0.083 |
|
|
|
| 10 |
H |
0.091 |
|
|
|
| 11 |
H |
0.079 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.835 |
1.418 |
0.350 |
2.346 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.711 |
0.624 |
0.222 |
| y |
0.624 |
-29.503 |
0.322 |
| z |
0.222 |
0.322 |
-30.174 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.872 |
0.624 |
0.222 |
| y |
0.624 |
1.939 |
0.322 |
| z |
0.222 |
0.322 |
0.933 |
|
| Polar |
|---|
| 3z2-r2 | 1.867 |
|---|
| x2-y2 | -3.207 |
|---|
| xy | 0.624 |
|---|
| xz | 0.222 |
|---|
| yz | 0.322 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.651 |
-0.621 |
-0.160 |
| y |
-0.621 |
3.110 |
0.150 |
| z |
-0.160 |
0.150 |
2.439 |
<r2> (average value of r
2) Å
2
| <r2> |
132.677 |
| (<r2>)1/2 |
11.519 |