Vibrational Frequencies calculated at PBEPBE/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3088 |
3059 |
5.74 |
|
|
|
| 2 |
A |
3071 |
3043 |
14.00 |
|
|
|
| 3 |
A |
3049 |
3021 |
18.96 |
|
|
|
| 4 |
A |
3027 |
3000 |
14.87 |
|
|
|
| 5 |
A |
3017 |
2990 |
7.94 |
|
|
|
| 6 |
A |
3005 |
2978 |
3.92 |
|
|
|
| 7 |
A |
2976 |
2949 |
21.76 |
|
|
|
| 8 |
A |
2971 |
2943 |
0.60 |
|
|
|
| 9 |
A |
1447 |
1434 |
3.92 |
|
|
|
| 10 |
A |
1441 |
1428 |
5.71 |
|
|
|
| 11 |
A |
1436 |
1423 |
8.37 |
|
|
|
| 12 |
A |
1422 |
1409 |
1.24 |
|
|
|
| 13 |
A |
1361 |
1349 |
7.83 |
|
|
|
| 14 |
A |
1343 |
1330 |
13.67 |
|
|
|
| 15 |
A |
1297 |
1285 |
7.13 |
|
|
|
| 16 |
A |
1281 |
1270 |
8.97 |
|
|
|
| 17 |
A |
1242 |
1231 |
17.67 |
|
|
|
| 18 |
A |
1228 |
1217 |
22.34 |
|
|
|
| 19 |
A |
1143 |
1133 |
8.09 |
|
|
|
| 20 |
A |
1095 |
1085 |
7.48 |
|
|
|
| 21 |
A |
1087 |
1077 |
5.83 |
|
|
|
| 22 |
A |
1039 |
1029 |
2.03 |
|
|
|
| 23 |
A |
1000 |
991 |
18.54 |
|
|
|
| 24 |
A |
931 |
922 |
3.20 |
|
|
|
| 25 |
A |
889 |
881 |
6.38 |
|
|
|
| 26 |
A |
762 |
755 |
10.72 |
|
|
|
| 27 |
A |
728 |
721 |
36.66 |
|
|
|
| 28 |
A |
596 |
591 |
32.48 |
|
|
|
| 29 |
A |
427 |
423 |
3.13 |
|
|
|
| 30 |
A |
396 |
393 |
5.55 |
|
|
|
| 31 |
A |
329 |
326 |
3.85 |
|
|
|
| 32 |
A |
248 |
246 |
0.40 |
|
|
|
| 33 |
A |
236 |
234 |
0.26 |
|
|
|
| 34 |
A |
146 |
145 |
2.25 |
|
|
|
| 35 |
A |
115 |
114 |
1.17 |
|
|
|
| 36 |
A |
73 |
73 |
3.37 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 24469.5 cm
-1
Scaled (by 0.9909) Zero Point Vibrational Energy (zpe) 24246.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
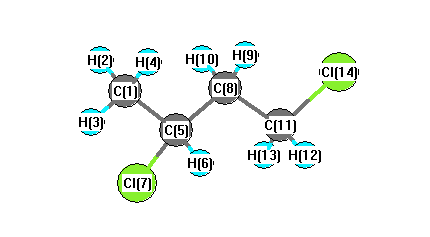
Charges from optimized geometry at PBEPBE/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.275 |
|
|
|
| 2 |
H |
0.144 |
|
|
|
| 3 |
H |
0.151 |
|
|
|
| 4 |
H |
0.131 |
|
|
|
| 5 |
C |
-0.342 |
|
|
|
| 6 |
H |
0.195 |
|
|
|
| 7 |
Cl |
-0.091 |
|
|
|
| 8 |
C |
-0.204 |
|
|
|
| 9 |
H |
0.157 |
|
|
|
| 10 |
H |
0.168 |
|
|
|
| 11 |
C |
-0.326 |
|
|
|
| 12 |
H |
0.181 |
|
|
|
| 13 |
H |
0.208 |
|
|
|
| 14 |
Cl |
-0.096 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.375 |
1.833 |
1.040 |
2.516 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-60.372 |
-3.934 |
0.082 |
| y |
-3.934 |
-51.802 |
0.072 |
| z |
0.082 |
0.072 |
-51.181 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-8.880 |
-3.934 |
0.082 |
| y |
-3.934 |
3.974 |
0.072 |
| z |
0.082 |
0.072 |
4.906 |
|
| Polar |
|---|
| 3z2-r2 | 9.812 |
|---|
| x2-y2 | -8.569 |
|---|
| xy | -3.934 |
|---|
| xz | 0.082 |
|---|
| yz | 0.072 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.831 |
0.373 |
-0.302 |
| y |
0.373 |
10.304 |
0.130 |
| z |
-0.302 |
0.130 |
7.868 |
<r2> (average value of r
2) Å
2
| <r2> |
372.443 |
| (<r2>)1/2 |
19.299 |