Jump to
S1C2
Energy calculated at PBEPBE/6-311G**
| | hartrees |
|---|
| Energy at 0K | -958.647071 |
| Energy at 298.15K | |
| HF Energy | -958.647071 |
| Nuclear repulsion energy | 124.965948 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3199 |
3170 |
0.02 |
|
|
|
| 2 |
A1 |
737 |
730 |
7.12 |
|
|
|
| 3 |
A1 |
309 |
306 |
0.08 |
|
|
|
| 4 |
B1 |
332i |
329i |
63.21 |
|
|
|
| 5 |
B2 |
1208 |
1197 |
41.70 |
|
|
|
| 6 |
B2 |
890 |
882 |
182.92 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3005.5 cm
-1
Scaled (by 0.9909) Zero Point Vibrational Energy (zpe) 2978.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
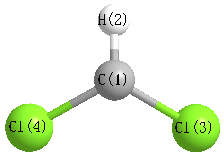
Geometric Data calculated at PBEPBE/6-311G**
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.671 |
| H2 |
0.000 |
0.000 |
1.755 |
| Cl3 |
0.000 |
1.487 |
-0.170 |
| Cl4 |
0.000 |
-1.487 |
-0.170 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
Cl3 |
Cl4 |
| C1 | | 1.0844 | 1.7079 | 1.7079 |
H2 | 1.0844 | | 2.4324 | 2.4324 | Cl3 | 1.7079 | 2.4324 | | 2.9733 | Cl4 | 1.7079 | 2.4324 | 2.9733 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Cl3 |
C1 |
H2 |
119.490 |
|
Cl3 |
C1 |
Cl4 |
121.020 |
| Cl4 |
C1 |
H2 |
119.490 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.320 |
|
|
|
| 2 |
H |
0.221 |
|
|
|
| 3 |
Cl |
0.050 |
|
|
|
| 4 |
Cl |
0.050 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.884 |
0.884 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.508 |
0.000 |
0.000 |
| y |
0.000 |
-31.354 |
0.000 |
| z |
0.000 |
0.000 |
-29.446 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-2.108 |
0.000 |
0.000 |
| y |
0.000 |
-0.377 |
0.000 |
| z |
0.000 |
0.000 |
2.485 |
|
| Polar |
|---|
| 3z2-r2 | 4.969 |
|---|
| x2-y2 | -1.154 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.265 |
0.000 |
0.000 |
| y |
0.000 |
7.351 |
0.000 |
| z |
0.000 |
0.000 |
3.770 |
<r2> (average value of r
2) Å
2
| <r2> |
101.334 |
| (<r2>)1/2 |
10.066 |
Jump to
S1C1
Energy calculated at PBEPBE/6-311G**
| | hartrees |
|---|
| Energy at 0K | -958.647720 |
| Energy at 298.15K | -958.648457 |
| HF Energy | -958.647720 |
| Nuclear repulsion energy | 124.744590 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3159 |
3130 |
1.92 |
|
|
|
| 2 |
A' |
742 |
735 |
12.54 |
|
|
|
| 3 |
A' |
439 |
435 |
35.92 |
|
|
|
| 4 |
A' |
303 |
300 |
0.90 |
|
|
|
| 5 |
A" |
1213 |
1202 |
34.39 |
|
|
|
| 6 |
A" |
857 |
849 |
208.35 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3356.0 cm
-1
Scaled (by 0.9909) Zero Point Vibrational Energy (zpe) 3325.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/6-311G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.012 |
0.694 |
0.000 |
| H2 |
-0.477 |
1.665 |
0.000 |
| Cl3 |
0.012 |
-0.171 |
1.484 |
| Cl4 |
0.012 |
-0.171 |
-1.484 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
Cl3 |
Cl4 |
| C1 | | 1.0878 | 1.7179 | 1.7179 |
H2 | 1.0878 | | 2.4115 | 2.4115 | Cl3 | 1.7179 | 2.4115 | | 2.9682 | Cl4 | 1.7179 | 2.4115 | 2.9682 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Cl3 |
C1 |
H2 |
116.729 |
|
Cl3 |
C1 |
Cl4 |
119.521 |
| Cl4 |
C1 |
H2 |
116.729 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.304 |
|
|
|
| 2 |
H |
0.218 |
|
|
|
| 3 |
Cl |
0.043 |
|
|
|
| 4 |
Cl |
0.043 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.543 |
0.791 |
0.000 |
0.960 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-32.219 |
-0.929 |
0.000 |
| y |
-0.929 |
-29.899 |
0.000 |
| z |
0.000 |
0.000 |
-31.531 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.504 |
-0.929 |
0.000 |
| y |
-0.929 |
1.976 |
0.000 |
| z |
0.000 |
0.000 |
-0.472 |
|
| Polar |
|---|
| 3z2-r2 | -0.943 |
|---|
| x2-y2 | -2.320 |
|---|
| xy | -0.929 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.354 |
-0.162 |
0.000 |
| y |
-0.162 |
3.802 |
0.000 |
| z |
0.000 |
0.000 |
7.445 |
<r2> (average value of r
2) Å
2
| <r2> |
101.277 |
| (<r2>)1/2 |
10.064 |