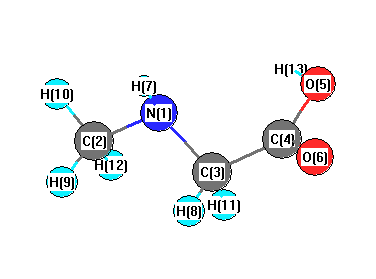
Vibrational Frequencies calculated at PBEPBE/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3442 |
3395 |
2.36 |
|
|
|
| 2 |
A |
3173 |
3129 |
383.90 |
|
|
|
| 3 |
A |
3074 |
3032 |
17.95 |
|
|
|
| 4 |
A |
3033 |
2991 |
1.52 |
|
|
|
| 5 |
A |
3030 |
2988 |
31.48 |
|
|
|
| 6 |
A |
2979 |
2938 |
18.60 |
|
|
|
| 7 |
A |
2936 |
2896 |
80.52 |
|
|
|
| 8 |
A |
1830 |
1805 |
320.88 |
|
|
|
| 9 |
A |
1481 |
1460 |
9.44 |
|
|
|
| 10 |
A |
1461 |
1441 |
37.47 |
|
|
|
| 11 |
A |
1445 |
1426 |
23.11 |
|
|
|
| 12 |
A |
1433 |
1413 |
41.34 |
|
|
|
| 13 |
A |
1417 |
1398 |
67.38 |
|
|
|
| 14 |
A |
1405 |
1386 |
237.19 |
|
|
|
| 15 |
A |
1291 |
1274 |
3.78 |
|
|
|
| 16 |
A |
1247 |
1230 |
2.76 |
|
|
|
| 17 |
A |
1195 |
1179 |
22.21 |
|
|
|
| 18 |
A |
1141 |
1125 |
29.60 |
|
|
|
| 19 |
A |
1118 |
1103 |
26.73 |
|
|
|
| 20 |
A |
1100 |
1085 |
10.87 |
|
|
|
| 21 |
A |
990 |
976 |
33.33 |
|
|
|
| 22 |
A |
946 |
933 |
25.50 |
|
|
|
| 23 |
A |
926 |
913 |
30.82 |
|
|
|
| 24 |
A |
852 |
841 |
14.50 |
|
|
|
| 25 |
A |
771 |
760 |
63.49 |
|
|
|
| 26 |
A |
625 |
616 |
3.40 |
|
|
|
| 27 |
A |
551 |
543 |
5.74 |
|
|
|
| 28 |
A |
462 |
455 |
7.92 |
|
|
|
| 29 |
A |
374 |
369 |
5.47 |
|
|
|
| 30 |
A |
288 |
284 |
6.25 |
|
|
|
| 31 |
A |
204 |
201 |
1.97 |
|
|
|
| 32 |
A |
141 |
139 |
2.42 |
|
|
|
| 33 |
A |
72 |
71 |
5.83 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23214.7 cm
-1
Scaled (by 0.9863) Zero Point Vibrational Energy (zpe) 22896.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.530 |
|
|
|
| 2 |
C |
-0.237 |
|
|
|
| 3 |
C |
-0.188 |
|
|
|
| 4 |
C |
0.526 |
|
|
|
| 5 |
O |
-0.452 |
|
|
|
| 6 |
O |
-0.431 |
|
|
|
| 7 |
H |
0.272 |
|
|
|
| 8 |
H |
0.148 |
|
|
|
| 9 |
H |
0.122 |
|
|
|
| 10 |
H |
0.138 |
|
|
|
| 11 |
H |
0.164 |
|
|
|
| 12 |
H |
0.146 |
|
|
|
| 13 |
H |
0.324 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
5.392 |
-0.340 |
-0.668 |
5.444 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.183 |
-0.590 |
-0.457 |
| y |
-0.590 |
-37.080 |
-0.588 |
| z |
-0.457 |
-0.588 |
-32.947 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-7.169 |
-0.590 |
-0.457 |
| y |
-0.590 |
0.485 |
-0.588 |
| z |
-0.457 |
-0.588 |
6.685 |
|
| Polar |
|---|
| 3z2-r2 | 13.370 |
|---|
| x2-y2 | -5.103 |
|---|
| xy | -0.590 |
|---|
| xz | -0.457 |
|---|
| yz | -0.588 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.717 |
0.434 |
0.007 |
| y |
0.434 |
6.748 |
0.023 |
| z |
0.007 |
0.023 |
5.203 |
<r2> (average value of r
2) Å
2
| <r2> |
194.161 |
| (<r2>)1/2 |
13.934 |