Vibrational Frequencies calculated at PBEPBE/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3102 |
3059 |
15.07 |
|
|
|
| 2 |
A |
3079 |
3037 |
27.65 |
|
|
|
| 3 |
A |
3063 |
3021 |
2.07 |
|
|
|
| 4 |
A |
3031 |
2990 |
14.51 |
|
|
|
| 5 |
A |
3020 |
2979 |
2.28 |
|
|
|
| 6 |
A |
2973 |
2932 |
22.08 |
|
|
|
| 7 |
A |
2963 |
2923 |
7.12 |
|
|
|
| 8 |
A |
2281 |
2250 |
8.28 |
|
|
|
| 9 |
A |
1705 |
1682 |
2.11 |
|
|
|
| 10 |
A |
1453 |
1433 |
10.09 |
|
|
|
| 11 |
A |
1441 |
1422 |
7.45 |
|
|
|
| 12 |
A |
1411 |
1392 |
9.49 |
|
|
|
| 13 |
A |
1374 |
1355 |
2.15 |
|
|
|
| 14 |
A |
1311 |
1293 |
1.52 |
|
|
|
| 15 |
A |
1293 |
1275 |
0.55 |
|
|
|
| 16 |
A |
1254 |
1237 |
6.13 |
|
|
|
| 17 |
A |
1185 |
1169 |
0.20 |
|
|
|
| 18 |
A |
1106 |
1091 |
0.42 |
|
|
|
| 19 |
A |
1061 |
1046 |
4.93 |
|
|
|
| 20 |
A |
1028 |
1014 |
0.12 |
|
|
|
| 21 |
A |
969 |
956 |
28.59 |
|
|
|
| 22 |
A |
927 |
914 |
10.55 |
|
|
|
| 23 |
A |
923 |
910 |
4.64 |
|
|
|
| 24 |
A |
874 |
862 |
0.28 |
|
|
|
| 25 |
A |
741 |
730 |
0.60 |
|
|
|
| 26 |
A |
551 |
544 |
0.82 |
|
|
|
| 27 |
A |
441 |
435 |
0.42 |
|
|
|
| 28 |
A |
371 |
366 |
0.39 |
|
|
|
| 29 |
A |
284 |
280 |
1.03 |
|
|
|
| 30 |
A |
257 |
253 |
4.67 |
|
|
|
| 31 |
A |
207 |
204 |
1.33 |
|
|
|
| 32 |
A |
133 |
131 |
3.41 |
|
|
|
| 33 |
A |
69 |
68 |
1.90 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22940.2 cm
-1
Scaled (by 0.9863) Zero Point Vibrational Energy (zpe) 22625.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
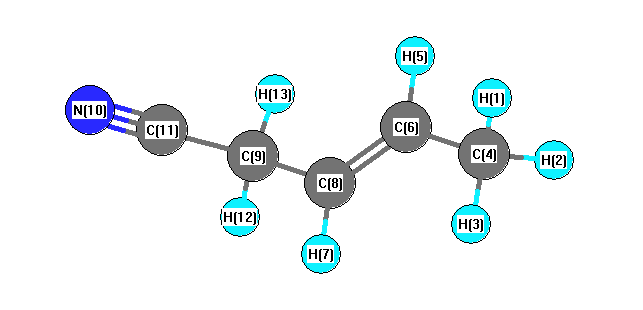
Charges from optimized geometry at PBEPBE/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.140 |
|
|
|
| 2 |
H |
0.144 |
|
|
|
| 3 |
H |
0.133 |
|
|
|
| 4 |
C |
-0.408 |
|
|
|
| 5 |
H |
0.099 |
|
|
|
| 6 |
C |
-0.062 |
|
|
|
| 7 |
H |
0.113 |
|
|
|
| 8 |
C |
-0.056 |
|
|
|
| 9 |
C |
-0.339 |
|
|
|
| 10 |
N |
-0.461 |
|
|
|
| 11 |
C |
0.348 |
|
|
|
| 12 |
H |
0.170 |
|
|
|
| 13 |
H |
0.179 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.578 |
2.153 |
0.288 |
4.186 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-44.726 |
-6.123 |
-1.669 |
| y |
-6.123 |
-36.360 |
-1.170 |
| z |
-1.669 |
-1.170 |
-34.640 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-9.226 |
-6.123 |
-1.669 |
| y |
-6.123 |
3.323 |
-1.170 |
| z |
-1.669 |
-1.170 |
5.903 |
|
| Polar |
|---|
| 3z2-r2 | 11.807 |
|---|
| x2-y2 | -8.366 |
|---|
| xy | -6.123 |
|---|
| xz | -1.669 |
|---|
| yz | -1.170 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.228 |
0.447 |
-0.804 |
| y |
0.447 |
6.812 |
-0.400 |
| z |
-0.804 |
-0.400 |
6.578 |
<r2> (average value of r
2) Å
2
| <r2> |
237.011 |
| (<r2>)1/2 |
15.395 |