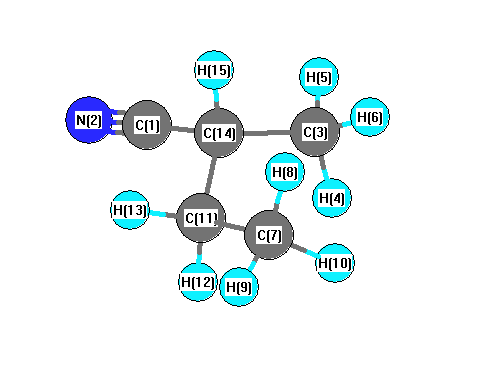
Vibrational Frequencies calculated at PBEPBE/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3090 |
3048 |
13.82 |
|
|
|
| 2 |
A |
3084 |
3041 |
15.19 |
|
|
|
| 3 |
A |
3073 |
3031 |
25.93 |
|
|
|
| 4 |
A |
3065 |
3023 |
31.29 |
|
|
|
| 5 |
A |
3032 |
2991 |
5.62 |
|
|
|
| 6 |
A |
3000 |
2959 |
13.23 |
|
|
|
| 7 |
A |
2991 |
2950 |
27.20 |
|
|
|
| 8 |
A |
2986 |
2945 |
9.82 |
|
|
|
| 9 |
A |
2960 |
2920 |
4.45 |
|
|
|
| 10 |
A |
2275 |
2243 |
9.14 |
|
|
|
| 11 |
A |
1475 |
1455 |
3.36 |
|
|
|
| 12 |
A |
1468 |
1448 |
8.12 |
|
|
|
| 13 |
A |
1464 |
1444 |
6.09 |
|
|
|
| 14 |
A |
1457 |
1437 |
9.10 |
|
|
|
| 15 |
A |
1448 |
1428 |
0.24 |
|
|
|
| 16 |
A |
1377 |
1358 |
2.22 |
|
|
|
| 17 |
A |
1369 |
1351 |
5.51 |
|
|
|
| 18 |
A |
1334 |
1316 |
0.36 |
|
|
|
| 19 |
A |
1303 |
1285 |
1.31 |
|
|
|
| 20 |
A |
1274 |
1257 |
2.67 |
|
|
|
| 21 |
A |
1243 |
1226 |
1.64 |
|
|
|
| 22 |
A |
1150 |
1135 |
2.57 |
|
|
|
| 23 |
A |
1108 |
1092 |
0.19 |
|
|
|
| 24 |
A |
1083 |
1068 |
5.42 |
|
|
|
| 25 |
A |
1029 |
1015 |
1.20 |
|
|
|
| 26 |
A |
972 |
959 |
2.42 |
|
|
|
| 27 |
A |
948 |
935 |
6.04 |
|
|
|
| 28 |
A |
882 |
870 |
0.91 |
|
|
|
| 29 |
A |
793 |
782 |
0.64 |
|
|
|
| 30 |
A |
748 |
738 |
4.02 |
|
|
|
| 31 |
A |
549 |
542 |
0.15 |
|
|
|
| 32 |
A |
524 |
517 |
1.03 |
|
|
|
| 33 |
A |
387 |
382 |
0.27 |
|
|
|
| 34 |
A |
318 |
314 |
0.46 |
|
|
|
| 35 |
A |
274 |
270 |
0.09 |
|
|
|
| 36 |
A |
222 |
219 |
0.15 |
|
|
|
| 37 |
A |
201 |
198 |
1.99 |
|
|
|
| 38 |
A |
157 |
155 |
4.81 |
|
|
|
| 39 |
A |
83 |
82 |
1.39 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 28097.1 cm
-1
Scaled (by 0.9863) Zero Point Vibrational Energy (zpe) 27712.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.347 |
|
|
|
| 2 |
N |
-0.474 |
|
|
|
| 3 |
C |
-0.359 |
|
|
|
| 4 |
H |
0.142 |
|
|
|
| 5 |
H |
0.144 |
|
|
|
| 6 |
H |
0.134 |
|
|
|
| 7 |
C |
-0.375 |
|
|
|
| 8 |
H |
0.126 |
|
|
|
| 9 |
H |
0.133 |
|
|
|
| 10 |
H |
0.129 |
|
|
|
| 11 |
C |
-0.209 |
|
|
|
| 12 |
H |
0.133 |
|
|
|
| 13 |
H |
0.137 |
|
|
|
| 14 |
C |
-0.165 |
|
|
|
| 15 |
H |
0.158 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.787 |
1.274 |
0.468 |
4.022 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.718 |
-3.737 |
-0.823 |
| y |
-3.737 |
-36.928 |
-0.109 |
| z |
-0.823 |
-0.109 |
-36.447 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-11.030 |
-3.737 |
-0.823 |
| y |
-3.737 |
5.155 |
-0.109 |
| z |
-0.823 |
-0.109 |
5.875 |
|
| Polar |
|---|
| 3z2-r2 | 11.751 |
|---|
| x2-y2 | -10.790 |
|---|
| xy | -3.737 |
|---|
| xz | -0.823 |
|---|
| yz | -0.109 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.636 |
0.514 |
0.259 |
| y |
0.514 |
7.973 |
0.116 |
| z |
0.259 |
0.116 |
6.914 |
<r2> (average value of r
2) Å
2
| <r2> |
200.780 |
| (<r2>)1/2 |
14.170 |