Jump to
S1C2
S2C1
S2C2
Energy calculated at PBEPBE/6-31G**
| | hartrees |
|---|
| Energy at 0K | -131.262116 |
| Energy at 298.15K | -131.261405 |
| HF Energy | -131.262116 |
| Nuclear repulsion energy | 46.900247 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3340 |
3294 |
36.98 |
|
|
|
| 2 |
A' |
1820 |
1795 |
23.59 |
|
|
|
| 3 |
A' |
1225 |
1208 |
3.94 |
|
|
|
| 4 |
A' |
466 |
460 |
28.04 |
|
|
|
| 5 |
A' |
382 |
377 |
24.80 |
|
|
|
| 6 |
A" |
438 |
432 |
1.91 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3835.3 cm
-1
Scaled (by 0.9863) Zero Point Vibrational Energy (zpe) 3782.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/6-31G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.067 |
-1.230 |
0.000 |
| C2 |
0.000 |
0.083 |
0.000 |
| N3 |
-0.144 |
1.291 |
0.000 |
| H4 |
0.609 |
-2.162 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3148 | 2.5299 | 1.0785 |
C2 | 1.3148 | | 1.2165 | 2.3264 | N3 | 2.5299 | 1.2165 | | 3.5344 | H4 | 1.0785 | 2.3264 | 3.5344 | |
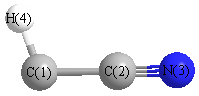 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
176.099 |
|
C2 |
C1 |
H4 |
152.703 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S2C1
S2C2
Energy calculated at PBEPBE/6-31G**
| | hartrees |
|---|
| Energy at 0K | -131.261518 |
| Energy at 298.15K | |
| HF Energy | -131.261518 |
| Nuclear repulsion energy | 46.947029 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
3399 |
3352 |
68.46 |
|
|
|
| 2 |
Σ |
1784 |
1760 |
24.08 |
|
|
|
| 3 |
Σ |
1254 |
1237 |
3.08 |
|
|
|
| 4 |
Π |
441 |
435 |
0.91 |
|
|
|
| 4 |
Π |
441 |
435 |
0.91 |
|
|
|
| 5 |
Π |
296i |
292i |
48.82 |
|
|
|
| 5 |
Π |
296i |
292i |
48.82 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3362.6 cm
-1
Scaled (by 0.9863) Zero Point Vibrational Energy (zpe) 3316.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/6-31G**
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.220 |
| C2 |
0.000 |
0.000 |
0.081 |
| N3 |
0.000 |
0.000 |
1.304 |
| H4 |
0.000 |
0.000 |
-2.293 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3006 | 2.5239 | 1.0736 |
C2 | 1.3006 | | 1.2232 | 2.3743 | N3 | 2.5239 | 1.2232 | | 3.5975 | H4 | 1.0736 | 2.3743 | 3.5975 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
180.000 |
|
C2 |
C1 |
H4 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.029 |
|
|
|
| 2 |
C |
0.294 |
|
|
|
| 3 |
N |
-0.467 |
|
|
|
| 4 |
H |
0.203 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-3.190 |
3.190 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-16.884 |
0.000 |
0.000 |
| y |
0.000 |
-16.884 |
0.000 |
| z |
0.000 |
0.000 |
-15.114 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.885 |
0.000 |
0.000 |
| y |
0.000 |
-0.885 |
0.000 |
| z |
0.000 |
0.000 |
1.770 |
|
| Polar |
|---|
| 3z2-r2 | 3.540 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.653 |
0.000 |
0.000 |
| y |
0.000 |
1.653 |
0.000 |
| z |
0.000 |
0.000 |
6.319 |
<r2> (average value of r
2) Å
2
| <r2> |
36.308 |
| (<r2>)1/2 |
6.026 |
Jump to
S1C1
S1C2
S2C2
Energy calculated at PBEPBE/6-31G**
| | hartrees |
|---|
| Energy at 0K | -131.234709 |
| Energy at 298.15K | -131.234103 |
| HF Energy | -131.234709 |
| Nuclear repulsion energy | 46.561958 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3002 |
2961 |
12.96 |
|
|
|
| 2 |
A' |
2028 |
2001 |
13.91 |
|
|
|
| 3 |
A' |
1097 |
1082 |
18.05 |
|
|
|
| 4 |
A' |
884 |
872 |
65.70 |
|
|
|
| 5 |
A' |
433 |
427 |
20.18 |
|
|
|
| 6 |
A" |
301 |
297 |
7.57 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3873.0 cm
-1
Scaled (by 0.9863) Zero Point Vibrational Energy (zpe) 3819.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at PBEPBE/6-31G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.322 |
-1.246 |
0.000 |
| C2 |
0.000 |
0.090 |
0.000 |
| N3 |
-0.479 |
1.192 |
0.000 |
| H4 |
1.420 |
-1.409 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3739 | 2.5665 | 1.1100 |
C2 | 1.3739 | | 1.2022 | 2.0648 | N3 | 2.5665 | 1.2022 | | 3.2210 | H4 | 1.1100 | 2.0648 | 3.2210 | |
More geometry information
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.103 |
|
|
|
| 2 |
C |
0.326 |
|
|
|
| 3 |
N |
-0.390 |
|
|
|
| 4 |
H |
0.166 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.896 |
-0.987 |
0.000 |
2.137 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-16.014 |
-1.400 |
0.000 |
| y |
-1.400 |
-21.049 |
0.000 |
| z |
0.000 |
0.000 |
-15.548 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
2.285 |
-1.400 |
0.000 |
| y |
-1.400 |
-5.268 |
0.000 |
| z |
0.000 |
0.000 |
2.983 |
|
| Polar |
|---|
| 3z2-r2 | 5.967 |
|---|
| x2-y2 | 5.035 |
|---|
| xy | -1.400 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.912 |
-1.484 |
0.000 |
| y |
-1.484 |
5.860 |
0.000 |
| z |
0.000 |
0.000 |
2.153 |
<r2> (average value of r
2) Å
2
| <r2> |
36.498 |
| (<r2>)1/2 |
6.041 |
Jump to
S1C1
S1C2
S2C1
Energy calculated at PBEPBE/6-31G**
| | hartrees |
|---|
| Energy at 0K | -131.234709 |
| Energy at 298.15K | -131.234103 |
| HF Energy | -131.234709 |
| Nuclear repulsion energy | 46.561958 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at PBEPBE/6-31G**
Geometric Data calculated at PBEPBE/6-31G**
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability