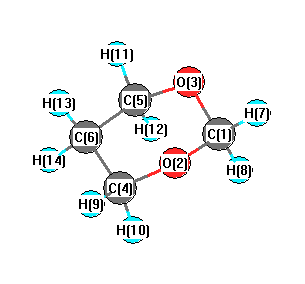
Vibrational Frequencies calculated at PBEPBE/6-31G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3063 |
3021 |
32.28 |
|
|
|
| 2 |
A' |
3052 |
3011 |
27.47 |
|
|
|
| 3 |
A' |
3042 |
3000 |
21.00 |
|
|
|
| 4 |
A' |
2992 |
2951 |
18.81 |
|
|
|
| 5 |
A' |
2896 |
2857 |
106.24 |
|
|
|
| 6 |
A' |
2834 |
2795 |
85.90 |
|
|
|
| 7 |
A' |
1466 |
1445 |
6.35 |
|
|
|
| 8 |
A' |
1452 |
1432 |
0.21 |
|
|
|
| 9 |
A' |
1427 |
1408 |
5.15 |
|
|
|
| 10 |
A' |
1368 |
1349 |
20.98 |
|
|
|
| 11 |
A' |
1282 |
1265 |
4.57 |
|
|
|
| 12 |
A' |
1169 |
1153 |
47.45 |
|
|
|
| 13 |
A' |
1141 |
1125 |
116.57 |
|
|
|
| 14 |
A' |
1082 |
1067 |
16.66 |
|
|
|
| 15 |
A' |
980 |
967 |
35.63 |
|
|
|
| 16 |
A' |
896 |
884 |
9.93 |
|
|
|
| 17 |
A' |
828 |
817 |
10.51 |
|
|
|
| 18 |
A' |
626 |
617 |
4.04 |
|
|
|
| 19 |
A' |
475 |
468 |
0.48 |
|
|
|
| 20 |
A' |
431 |
425 |
8.99 |
|
|
|
| 21 |
A' |
265 |
261 |
1.74 |
|
|
|
| 22 |
A" |
3043 |
3001 |
43.19 |
|
|
|
| 23 |
A" |
2888 |
2849 |
21.33 |
|
|
|
| 24 |
A" |
1452 |
1432 |
5.30 |
|
|
|
| 25 |
A" |
1398 |
1379 |
13.42 |
|
|
|
| 26 |
A" |
1344 |
1325 |
0.44 |
|
|
|
| 27 |
A" |
1323 |
1305 |
0.19 |
|
|
|
| 28 |
A" |
1293 |
1275 |
0.97 |
|
|
|
| 29 |
A" |
1218 |
1201 |
24.09 |
|
|
|
| 30 |
A" |
1191 |
1175 |
0.36 |
|
|
|
| 31 |
A" |
1031 |
1017 |
17.15 |
|
|
|
| 32 |
A" |
994 |
980 |
108.71 |
|
|
|
| 33 |
A" |
895 |
882 |
25.84 |
|
|
|
| 34 |
A" |
873 |
861 |
0.05 |
|
|
|
| 35 |
A" |
451 |
445 |
6.38 |
|
|
|
| 36 |
A" |
262 |
258 |
1.44 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 26209.8 cm
-1
Scaled (by 0.9863) Zero Point Vibrational Energy (zpe) 25850.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at PBEPBE/6-31G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.200 |
|
|
|
| 2 |
O |
-0.423 |
|
|
|
| 3 |
O |
-0.423 |
|
|
|
| 4 |
C |
-0.011 |
|
|
|
| 5 |
C |
-0.011 |
|
|
|
| 6 |
C |
-0.236 |
|
|
|
| 7 |
H |
0.126 |
|
|
|
| 8 |
H |
0.081 |
|
|
|
| 9 |
H |
0.126 |
|
|
|
| 10 |
H |
0.098 |
|
|
|
| 11 |
H |
0.126 |
|
|
|
| 12 |
H |
0.098 |
|
|
|
| 13 |
H |
0.138 |
|
|
|
| 14 |
H |
0.113 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.575 |
1.781 |
0.000 |
1.871 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.897 |
1.627 |
0.000 |
| y |
1.627 |
-34.837 |
0.000 |
| z |
0.000 |
0.000 |
-38.390 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.716 |
1.627 |
0.000 |
| y |
1.627 |
2.307 |
0.000 |
| z |
0.000 |
0.000 |
-3.023 |
|
| Polar |
|---|
| 3z2-r2 | -6.047 |
|---|
| x2-y2 | -1.060 |
|---|
| xy | 1.627 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.756 |
0.510 |
0.000 |
| y |
0.510 |
7.845 |
0.000 |
| z |
0.000 |
0.000 |
7.309 |
<r2> (average value of r
2) Å
2
| <r2> |
140.445 |
| (<r2>)1/2 |
11.851 |